Der Pharma Chemica
Journal for Medicinal Chemistry, Pharmaceutical Chemistry, Pharmaceutical Sciences and Computational Chemistry
ISSN: 0975-413X
Abstract
A theoretical study of the inhibition of human 4-hydroxyphenylpyruvate dioxygenase by a series of pyrazalone-quinazolone hybrids
Author(s): Juan S. Gómez-Jeria* and Camila Moreno-RojasA Density Functional Theory study was carried out to find relationships between the electronic/molecular structure of a group of pyrazalone-quinazolone hybrids and their inhibition of human 4-hydroxyphenylpyruvate dioxygenase (HPPD). The geometries were fully optimized at the B3LYP/6-31G(d,p) level. A statistically significant equation was obtained. The equivalent 2D pharmacophore was built and some atom-site interactions were suggested. The analysis of the equation and the pharmacophore should provide new information about possible substitution sites for an enhancing of the inhibitory activity.
Select your language of interest to view the total content in your interested language

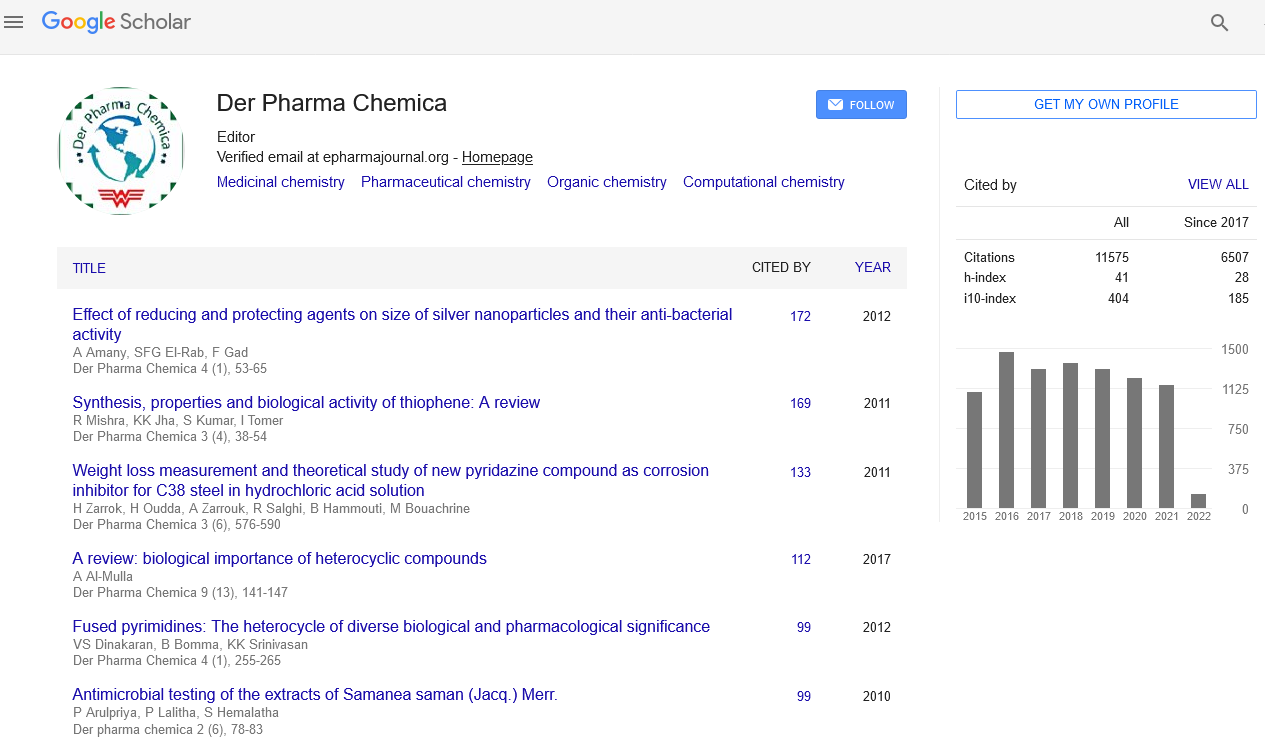
Google Scholar citation report
Citations : 25868
Der Pharma Chemica received 25868 citations as per Google Scholar report
Der Pharma Chemica peer review process verified at publons

DOWNLOADS
