Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 2
Synthesis, Antitubercular Activity and DNA-binding Study of some 2-Amino-3-cyano-4H-chromen-4-ylphosphonates
Gulzar A Khan1, Gowhar A Naikoo2, Javeed A War1, Umar Jan Pandit1, Mehraj Ud Din Sheikh1, Imran Khan1 and Ratnesh Das1*
1Department of Chemistry, Dr. Harisingh Gour Central University, Sagar, India
2Department of Mathematics and Sciences, College of Arts and Applied Sciences, Dhofar University, Sultanate of Oman
- *Corresponding Author:
- Ratnesh Das
Department of Chemistry
Dr. Harisingh Gour Central University
Sagar, India
Abstract
(2-Amino-3-cyano-4H-chromen-4-yl)-phosphonates 4a-e were synthesized on reacting substituted salicylaldehydes, malononitrile, and diethylphosphite via ultrasound irradiation in presence of catalytic amount of 4-Dimethylaminopyridine (DMAP). This protocol is an environmentally benign preparation and gives excellent yields of the target compounds (88-97%). The structures of new compounds were elucidated by spectroscopic techniques, including Fourier Transform Infra-Red (FTIR), Proton (1H-NMR), Carbon-13 (13C-NMR), Phosphorus- 31 (31P-NMR) Nuclear Magnetic Resonance and Liquid Chromatography/High Resolution Mass Spectrometry (LC-HRMS). All the compounds were screened for antitubercular activity against Mycobacterium tuberculosis H37 Rv using disc diffusion susceptibility method. The in vitro results indicate that all the compounds are potent. Among these 4d bearing nitro group at position C-2' is most effective and, gave a Minimal Inhibitory Concentration (MIC) value of 10 μg/ml in reference with Isoniazid at the same concentrations. DNA binding study of compound 4d with salmon milt DNA (sm-DNA) was confirmed with UV-Visible, fluorescence, cyclic voltammetry and molecular docking. It was revealed that 4d has a strong affinity towards sm-DNA and binds at DNA minor groove with a binding constant (K) 5.317 × 105 mol-1. Molecular docking calculations predicted strong affinity of 4d towards sm-DNA with a binding affinity (ΔG) -7.4 kcal mol-1. Non-covalent interactions, H-bonding and van der Waals forces were predicted as the driving forces of interaction. The compound 4d exhibited selective affinity towards adeninethiamine (A-T) base pairs.
Graphical Abstract
Keywords
Chromenyl phosphonates, Ultrasound, Antitubercular activity, DNA binding study, Molecular docking
| HIGHLIGHTS ➢ Ultrasonic-assisted green synthesis of (2-Amino-3-cyano-4H-chromen-4-yl)-phosphonates 4a-e. ➢ Using of both 4-DMAP and ultrasound irradiation for improvement of yields and reaction times than the reported methods. ➢ In vitro disc diffusion susceptibility test against Mycobacterium tuberculosis H37 Rv gave highly promising results. DNA-binding study of the most potent molecule 4d with sm-DNA under simulated physiological pH was probed with UV-Visible absorption, fluorescence quenching, cyclic voltammetry and molecular docking techniques. |
Introduction
Tuberculosis (TB) is considered an intractable disease and as such a major cause of death worldwide. On an average about 100 million people become affected by TB annually, and that more than 3 million people die each year from tuberculosis. As a consequence, it accounts about 3 million deaths globally every year [1]. The alarming increase in the number of TB cases today is its resurgence greatly dependent on two factors; development of TB 100-fold among HIV infected patients and drug resistance by some bacterial strains. However, some researchers have reported the drug resistance to as many as nine drugs [2]. Primary anti-TB drugs, such as Isonicotinic Acid Hydrazide (INH) in combination with streptomycin, and ethambutol, have been ineffective in dealing with some bacterial strains. These strains have evolved into Extreme Drug Resistant (XDR-TB) and Multidrug Resistant (MDR-TB) virulent forms due to partial or inadequate drug therapy. The synergism of the above two factors with HIV is an emerging threat which requires immediate attention [3]. The terrible situation has prompted WHO to declare TB a major global killer [4]. Antitubercular drugs acts by inhibiting the cell wall growth of target bacterial strains [5]. These drugs block the biosynthesis of type II mycoloic fatty acids (a vital component in bacterial cell walls) by controlling the activity of specific enzymes (e.g., Nicotinamide Adenine Dinucleotide (NADH) enoylase-reductase) necessary for cell wall formation [6].
Organphosphorus Compounds (OPCs) are frequently used in terms of agricultural, medicinal, and industrial applications [7]. Phosphonates a special class of OPC have been reported to possess paramount biological importance, including antibiotics, enzyme inhibitors and metabolic probes, pharmacological agents and peptide mimetics [8,9]. The last decade has witnessed tremendous development in exploring transformations for C-P bond formation [10]. In spite of so many methods, development of more efficient and superior protocols is still at great demand. The syntheses of OPCs have been reported via Pudovik, Kabaschnik-Field, Michaelis-Arbuzov, and phospha-Michael transformations [10-12]. Among these, phospha-Michael reaction involving the nucleophilic phosphite addition across carbon-carbon multiple bond is highly preferred and ever green method in C-P bond formation.
The development of Multicomponent Reactions (MCRs) is considered as one of the important future directions for carrying out sustainable organic transformations with fundamentally safer designs. MCRs facilitate molecular diversity, generated on mixing simply reaction precursors by facile creation of several new bonds in a single-step transformation with no need for intermediate isolation; their purification results in a bioactive heterocyclic product [5,13,14]. The use of MCRs to drive and expedite chemical transformations has made and continues to make a green impact in the field of synthetic organic chemistry, a fact well documented in the literature. High atom-economy, structural complexity, operational simplicity, and environmentally benign like advantageous features makes MCRs as powerful synthetically efficient tools in the hands of chemists to design and to develop a vast library of complex heterocyclic scaffolds [15-17].
Recently, there has been a growing interest in using sonochemistry as a safe, green and eco-compatible source of energy for the preparation bioactive heterocyclic scaffolds [9]. The prominent features of ultrasound are enhanced reaction rates, formation of purer products in high yields, easier work-up procedures, increased selectivity and significant reduction in time, solvent and waste products [18,19]. Such an additional convenience due to sonochemistry in the field of synthetic organic chemistry is related to cavitation (An origin in sonochemistry or a physical event that creates, enlarges and implodes gaseous and vaporous cavities in an irradiated liquid, thereby generating high temperatures and pressures accompanied by molecular fragmentation, ultimately results in the enhanced mass transfer and allowing chemical reactions to occur) [20].
The interaction of drugs with DNA has been widely studied for their application as probes for DNA structure and potential usage in chemotherapy. Drugs bind with DNA either by covalent or non-covalent mode. In non-covalent interaction (A case with majority of the synthetic organic molecules) the molecules bind with DNA in three ways: via, intercalation, groove binding or electrostatic interaction [21,22]. UV-Visible, fluorescence, cyclic voltammetry and molecular modelling techniques were used to study the DNA strand scission.
Thus, in continuation of our earlier work on TB [5,17], and DNA binding study [23,24] herein we report the antitubercular and DNA cleavage studies of 2-Amino-4H-chromen-4-ylphosphonates 4a-e.
Experimental Section
Chemicals and apparatus
Sigma-Aldrich, CDH and Merck purchased chemicals were used without purification to carry out this work. Melting points were determined using open capillary tube melting point apparatus and are presented without any correction. The Infra-Red (IR) spectra were recorded on a Fourier Transform Infra-Red (FTIR) Shimadzu-8400S spectrometer using KBr pellets. The Proton (1H-NMR), Carbon-13 (13C-NMR), Phosphorus-31 (31P-NMR) Nuclear Magnetic Resonance spectra’s were recorded on Bruker Avance 400 spectrometer using Tetramethylsilane (TMS) as the internal standard and CDCl3 as solvent. HR-LCMS were determined on Bruker Microtoff-QII 10330 mass spectrometer. The purity of the compounds was checked by Thin-Layer Chromatography (TLC) on F254 silica-gel pre coated sheets (Merck, Germany) which were visualized under UV light (254 and 365 nm), chloroform: methanol (7:2 v/v) as solvent systems.
General one pot synthesis of (2-Amino-3-cyano-4H-chromen-4-yl)-phosphonates (4a-e)
A mixture of equimolar amounts of substituted salicylaldehyde, malononitrile and diethylphosphite (1 mmol each) in aqueous ethanol (5 ml) containing 4-Dimethylaminopyridine (DMAP) (10 mol %) was irradiated at 25°C (The temperature inside the reactor was also 25°C) on plate of an ultrasonic cleaner at a frequency of 22 kHz for some time (Reaction complete based on TLC analysis)." The solid compound obtained was filtered off, washed with ethanol and recrystallized from suitable solvent to furnish the desired pure product (Scheme 1).
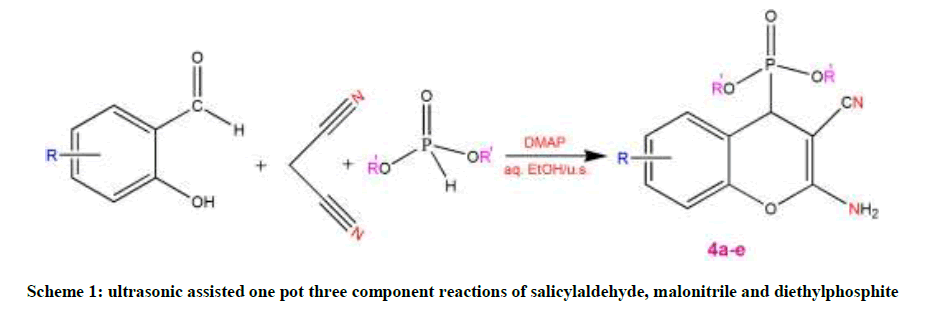
Scheme 1: ultrasonic assisted one pot three component reactions of salicylaldehyde, malonitrile and diethylphosphite
(2-Amino-6-chloro-3-cyano-4H-chromen-4-yl)-phosphonic acid diethyl ester (4a): FTIR (KBr) (Ѵmax cm-1): 3107 (C-H), 2178 (C≡N), 1663 (C=C), 1227 (P=O), 1087 (C−O−C), 974 (P−C), 869 (C-Cl); 1H-NMR (400 MHZ, CDCl3, TMS, δppm): 1.2 (t, J=7.2 Hz, 6H, CH3), 2.5 (q, J=6.0 Hz, 4H, CH2), 3.8 (H-9, d, J=18.4 Hz, 1H), 4.3 (brs, 2H, NH2), 7.0 (H-3, d, J=8.8 Hz, 1H), 7.3 (H-6, s, 1H), 7.4 (H-2, d, J=4.4 Hz, 1H); 13CNMR (100 MHZ, CDCl3, δppm) δc: 15.2 (C-12), 35.3 (C-9), 53.9 (C-11), 63.9 (C-8), 118.8 (C-10), 127.9 (C-3), 128.7 (C-2), 129.8 (C-5), 133.9 (C-1), 144.1 (C-6), 152.3 (C-4), 160.7 (C-7); 31P-NMR (161 MHz, CDCl3, δppm): 22.7; HR-LCMS (ESI): m/z [M]+: 342.0136.
(2-Amino-6-bromo-3-cyano-4H-chromen-4-yl)-phosphonic acid diethyl ester (4b): FTIR (KBr) (Ѵmax cm-1): 3103 (C-H), 2182 (C≡N), 1654 (C=C), 1436 (P−C), 1229 (P=O), 1089 (C−O−C), 971 (P−C), 635 (C-Br); 1H-NMR (400 MHZ, CDCl3, TMS, δppm): 1.2 (t, J=7.2 Hz, 6H, CH3), 2.5 (q, J=6.4 Hz, 4H, CH2), 3.8 (H-9, d, J=18.4 Hz, 1H), 4.3 (brs, 2H, NH2), 7.0 (H-3, d, J=8.8 Hz, 1H), 7.3 (H-6, s, 1H), 7.4 (H-2, d, J=4.0 Hz, 1H); 13C-NMR (100 MHZ, CDCl3, δppm) δc: 16.1 (C-12), 35.2 (C-9), 51.9 (C-8), 63.3 (C-11), 117.1 (C-10), 117.3 (C-1), 118.2 (C-3), 118.6 (C- 5), 131.9 (C-2), 132.2 (C-6), 148.3 (C-4), 161.5 (C-7); 31P-NMR (161 MHz, CDCl3, δppm): 20.7; HRLCMS (ESI): m/z [M]+: 385.9831.
(2-Amino-3-cyano-4H-chromen-4-yl)-phosphonic acid diethyl ester (4c): FTIR (KBr) (Ѵmax cm-1): 3097 (C-H), 2181 (C≡N), 1658 (C=C), 1443 (P−C), 1237 (P=O), 1077 (C−O−C), 963 (P−C); 1H-NMR (400 MHZ, CDCl3, TMS, δppm): 1.2 (t, J=6.8 Hz, 6H, CH3), 2.5 (q, J=6.0 Hz, 4H, CH2), 4.0 (H-9, d, J=18.8 Hz, 1H), 4.3 (brs, 2H, NH2), 7.0 (H-3, d, J=7.6 Hz, 1H), 7.3 (H-1, dd, J1=4.8 Hz & J2=4.0 Hz, 1H), 7.4 (H-6, d, J=7.2 Hz, 1H), 7.4 (H-2, dd, J1=4.8 Hz & J2=5.2 Hz, 1H); 13C-NMR (100 MHZ, CDCl3, δppm) δc: 16.4 (C-12), 35.3 (C-9), 51.8 (C-8), 63.2 (C- 11), 116.3 (C-3), 119.3 (C-10), 124.8 (C-6), 128.7 (C-1), 129.3 (C-5), 129.6 (C-2), 149.5 (C-4), 161.1 (C-7); 31P-NMR (161 MHz, CDCl3, δppm): 21.6; HR-LCMS (ESI): m/z [M]+: 308.0726.
(2-Amino-3-cyano-7-nitro-4H-chromen-4-yl)-phosphonic acid diethyl ester (4d): FTIR (KBr) (Ѵmax cm-1): 3123 (C-H), 2185 (C≡N), 1672 (C=C), 1448 (P−C), 1377 (N−O), 1239 (P=O), 1105 (C−O−C), 983 (P−C); 1H-NMR (400 MHZ, CDCl3, TMS, δppm): 1.2 (t, J=8.0 Hz, 6H, CH3), 2.5 (q, J=6.8 Hz, 4H, CH2), 3.8 (H-9, d, J=17.6 Hz, 1H), 4.7 (brs, 2H, NH2), 6.8 (H-6, d, J=8.0 Hz, 1H), 7.3 (H-3, s, 1H), 7.4 (H-1, d, J=4.4 Hz, 1H); 13C-NMR (100 MHZ, CDCl3, δppm) δc: 16.2 (C-12), 36.3 (C-9), 53.9 (C-8), 63.6 (C-11), 117.1 (C-3), 117.7 (C-10), 118.1 (C-1), 124.6 (C-6), 125.5 (C-5), 144.7 (C-2), 153.8 (C-4), 161.2 (C-7); 31P-NMR (161 MHz, CDCl3, δppm): 21.5; HR-LCMS (ESI): m/z [M]+: 353.0577.
(2-Amino-3-cyano-6-methyl-4H-chromen-4-yl)-phosphonic acid diethyl ester (4e): FTIR (KBr) (Ѵmax cm-1): 3119 (C-H), 2173 (C≡N), 1651 (C=C), 1456 (P−C), 1227 (P=O), 1137 (C−C), 1080 (C−O−C), 974 (P−C); 1H-NMR (400 MHZ, CDCl3, TMS, δppm): 1.2 (t, J=6.4 Hz, 6H, CH3), 2.1 (H-11, s, 3H), 2.5 (q, J=6.0 Hz, 4H, CH2), 3.0 (H-9, d, J=18.8 Hz, 1H), 4.7 (brs, 2H, NH2), 6.4 (H-3, d, J=8.4 Hz, 1H), 7.2 (H-6, s, 1H), 7.3 (H-2, d, J=3.6 Hz, 1H); 13C-NMR (100 MHZ, CDCl3, δppm) δc: 17.4 (C-13), 20.7 (C-11), 33.1 (C-9), 53.0 (C-8), 60.8 (C-12), 116.3 (C- 3), 117.2 (C-10), 127.9 (C-5), 128.3 (C-2), 142.5 (C-6), 154.6 (C-4), 159.8 (C-7); 31P-NMR (161 MHz, CDCl3, δppm): 21.9; HRLCMS (ESI): m/z [M]+: 321.9761.
In vitro biological evaluation assay
Antitubercular activity
Antitubercular activity was carried out by the disc diffusion susceptibility method [25]. Mycobacterium tuberculosis (MTCC, 300) purchased from the Institute of Microbial Technology (MTCC), Chandigarh (India) was cultured in blood nutrient agar medium in late logarithmic (A600 nm=1) fashion. Bacterial strains were cultured in the same media. It was shortly followed by streaking of 0.5 μl bacterial spread on LB agar plates (25 ml agar medium ± 90 μg/ml Isoniazid discs over 9 cm petri plates) as control. Filter discs (5 mm diameter) of Whatman range were treated with 5 μl of compound solutions including reference. After this, discs were air-dried for 7-10 min and kept over the plates. These plates were incubated at 37°C for about 48 h in a humid chamber. Following this bacterial zone-inhibition diameters were observed and measured carefully.
DNA-binding studies
Chemicals and reagents
A stock solution (5.5 × 10-3 mol.l-1) of the compound 4d was prepared in pH 7.4 phosphate buffer solution. A series of solutions was made through serial dilution for further use in UV-Visible and fluorescence titrations. Salmon milt DNA (sm-DNA) sodium salt was purchased from HiMedia and its stock solution (50 μM) was prepared by dissolving the required amount in pH 7.4 phosphate buffer solution. Its concentration (2.3 × 10-5) was identified spectrophotometrically using the molar absorption coefficient ε277=6600 M-1 cm-1.
Apparatus and measurements
UV-V is absorption measurements: UV-Visible absorption spectra were recorded on a Systronics double beam UV-Visible spectrophotometer- 2201. Two millilitres of 4d (5.5 × 10-3 mol.l-1) were titrated with increasing concentrations of 1 ml sm-DNA (0-45 μM). The solution was stirred and allowed to equilibrate for 10 min prior to recording the UV–visible spectra in the range 220-340 nm. To minimize experimental errors, baseline correction was performed prior to each recording.
Fluorescence measurements: Fluorescence spectral titrations were recorded on Shimadzu RF 5301 spectrofluorimetry equipped with a Xe lamp as the excitation source. Two millilitres of 4d (5.5 × 10-3 mol.l-1) were titrated with increasing concentrations of 1 ml sm-DNA (0-45 μM). The solution was stirred and allowed to equilibrate before recording the fluorescence emission spectra over a wavelength range of 250-800 nm with an excitation wavelength of 240 nm. The emission and excitation slit widths were adjusted to 10 nm each.
Electrochemical studies
The voltammetric study of compound 4d and its interaction with sm-DNA was performed at ambient temperature of 298 K (25°C) on a computer controlled NOVA software version 1.10.1.9 Metrohm Autolab B.V. PGSTAT128N equipped with a conventional three electrode system consisting of an Ag/AgCl (Saturated KCl) reference electrode, platinum wire as counter electrode and Glassy Carbon Electrode (GCE) as working electrode. A Systronics digital μ pH meter model-361 was used for pH measurements. For Cyclic Voltammetry (CV) solutions were prepared by mixing 7.0 ml of stock solution (5.5 × 10-3 mol.l-1) and 1.0 ml of 0.1 M LiCl (as supporting electrolyte) and 2.0 ml of 0.1 M phosphate buffer. Nitrogen gas was passed in the solution for ~15 min and thereafter, a blanket of nitrogen gas was maintained throughout the experiment.
Molecular docking
Molecular docking calculations were performed with Autodock Vina software [26]. The docking protocol used had previously been successful in predicting the exact experimental crystal conformation of co-crystallized inhibitors [24]. The crystal structure 1BNA was selected for docking all the synthesized compounds because it contains all four bases (A, T, G and C) in addition both minor and major grooves. Autodock Tools (ADT) was used to prepare DNA and ligands. Atomic charges for DNA were calculated using Kollman method, polar hydrogens were added and grid dimensions (40Å × 40Å × 40Å) were defined so to include both minor and major grooves of DNA. Genetic algorithm available in ADT was employed for docking. By default ligand was considered flexible and DNA as rigid. Another protocol was employed in docking in which AM1- BCC charges were added both on ligands and 1BNA. To check the comparative affinity of compound 4d towards A/T and C/G base pairs; crystal structures of six fragments of double stranded DNA (PDB IDs: 2ELG, 463D, 1VTJ, 1BNA, 1G3X, 1Z3F) [27-32] which differ in adenine-thiamine base pair content, were downloaded from the RCSB PDB website (www.rscb.org/pdb). Crystal structure of DNA (PDB ID: 1G3X) co-crystallized with acridine-peptide drug was utilized to check the accuracy of docking protocol. As is evident from Figure 1, the docking protocol we employed reproduced the crystal structure conformation with RMSD values within the reliable limit of 2Å [33].
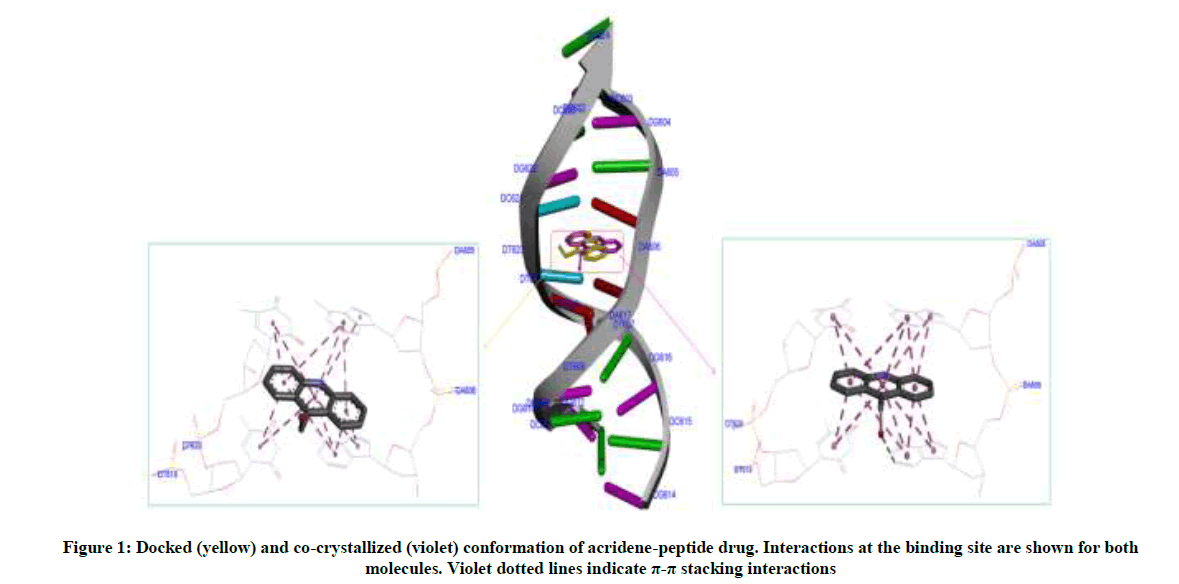
Figure 1: Docked (yellow) and co-crystallized (violet) conformation of acridene-peptide drug. Interactions at the binding site are shown for both molecules. Violet dotted lines indicate π-π stacking interactions
The ligands were prepared by converting 2D geometry to 3D geometry followed by optimization at B3LYP-631G (d,p) level of theory with Gaussian 09 software [34]. Atomic charges were calculated by Geistenger method. Torsions and rotatable bonds were defined in ADT. Autodock vina predicts bound conformations and calculates binding affinity. Both parameters have significance while carrying virtual screening. Out of the nine conformations predicted by Autodock Vina for each molecule, the highest scored conformation was visualized in DSV (Accelrys Software Inc.) and Pymol molecular graphic program (available at http://pymol.sourceforge.net) to check binding mode and interactions involved. The computations were done on an Intel core i3 2.20 GHz processor with 4 GB RAM.
Result and Discussion
Synthesis
In order to accomplish best experimental conditions, the reaction conditions were optimized for the preparation of 4a chosen as model reaction for both under conventional conditions and under ultrasound irradiation. Choice of solvent plays a significant role in many MCRs. Keeping optimal conditions in mind; reaction was initially carried out in solvents like Dichloroethane (DCE) and Dimethyl Formamide (DMF) in presence of the catalysts like anhydrous FeCl3 and Cs2CO3. It was observed that the product 4a was furnished with poor yield including prolonged reaction time (Table 1; Entry 1, 2; 9%, 23%) [35,36]. Afterwards, reaction was performed in solvents like Methanol (MeOH) and Ethanol (EtOH) using different catalysts viz, Et3N, Pyrrolidine, Ca(OH)2, K3PO4, Li(OH)2, and dibutylamine. The corresponding product yield was slightly improved at 25%, 38%, 46%, 34% and 70% as mentioned in Table 1 (Entry 3, 4, 5, 6, 7, 8); however prolonged reaction time were still observed [20,37-41]. Inspired by these results the reaction was then carried out in more polar aqueous medium using catalysts mpCuO and BiCl3. In this context, the reaction occurred smoothly and improved product yield as well as reaction time. As a result we were able to isolate the product yield at 73% and 80% (Table 1; Entry 9, 10) [5,18]. Finally, aqueous ethanol (EtOH/H2O) as a best solvent choice with DMAP as catalyst afforded 4a (Table 1; entry 11) in highest yield (96%) under ultrasound irradiation than all other solvents. The present strategy is robust in the presence of electron withdrawing and electron donating groups affording desired products with excellent yields (> 96%) in just a couple of minutes (Table 2).
| Entry | Solvent | Catalyst | Time (min) | % Yield |
|---|---|---|---|---|
| 1 | DCE | Anh. FeCl3 | P 95 Q 23 |
nrS 9 |
| 2 | DMF | Cs2 CO3 | P 85 Q 19 |
17 23 |
| 3 | MeOH | Et3N | P 81 Q 24 |
21 25 |
| 4 | MeOH:CH2Cl2 | Pyrrolidine | P 63 Q 14 |
nrS nrS |
| 5 | MeOH | Ca(OH)2 | P 61 Q 10 |
30 38 |
| 6 | EtOH | K3PO4 | P 43 Q 14 |
39 46 |
| 7 | EtOH | Li(OH)2 | P 55 Q 21 |
30 34 |
| 8 | EtOH | Dibutylamine | P 43 Q 13 |
63 70 |
| 9 | H2O | mpCuO | P 41 Q 8 |
65 73 |
| 10 | H2O | BiCl3 | P 31 Q 11 |
76 80 |
| 11 | EtOH/H2O | DMAP | Q 7 | 96 |
PConventional method; QSonochemical method; SNo reaction
Table 1: Optimized catalyst (or additives) and solvent effects on the reaction rate and % yield of (2-Amino-6-chloro-3-cyano-4H-chromen-4-yl)- phosphonic acid diethyl ester (4a)
| Entry | R | R'ˈ | Protocol | Time (min) | Product | (%) Isolated yield | m.p. (°C) |
|---|---|---|---|---|---|---|---|
| 1 | Cl | C2H5 | ultrasound | 7 | 4a | 96 | 152-154 |
| 2 | Br | C2H5 | ultrasound | 11 | 4b | 97 | 182-184 |
| 3 | H | C2H5 | ultrasound | 9 | 4c | 96 | 137-139 |
| 4 | NO2 | C2H5 | ultrasound | 9 | 4d | 88 | 130-132 |
| 5 | CH3 | C2H5 | ultrasound | 13 | 4e | 95 | 173-177 |
Table 2: Synthesis of (2-Amino-3-cyano-4H-chromen-4-yl)-phosphonates 4a-e
The synthesized compounds were structurally elucidated from spectral techniques including FTIR, 1H-NMR, 13C-NMR, 31P-NMR and LC-HRMS spectra. FT-IR spectrum of compound 4a exhibited sharp bands at 869 cm-1 and 974 cm-1 assigned to C-Cl and P−C is stretching frequencies respectively. The band at 2178 cm-1 indicated the presence of C≡N bond whereas aromatic C-H stretching was observed at 3107 cm- 1. In IH NMR spectrum of 4a a multiplet due to ethoxy protons were recorded at 1.2-2.5 ppm. The proton at C9ˈ position shows a doublet at 3.8 ppm whereas resonance emission of amine protons appeared as a broad singlet at 4.3 ppm. The rest aromatic proton signals were recorded in the expected range of 7.0-7.4 ppm. Also 13C NMR of compound 4a agrees with the number of carbons. It exhibited signals at 15.2 and 53.9 ppm attributed to ethoxy carbon atoms. A doublet at 35.3 ppm confirms the presence of a methine carbon directly attached to the phosphorus atom. Peaks between 127-144 ppm were assigned to aromatic carbon atoms. In the 31P NMR spectrum, a signal was recorded at 22.7 ppm. The above results were further supported by mass spectrum exhibited the molecular ion peak m/z 342.0136 [M]+, in agreement with the molecular weight of the compound.
In vitro antitubercular activity
As is evident from Table 3, compound 4a, 4b, and 4c have shown moderate growth inhibitory activities with respective MIC values of 20, 20, and 60 μg/ml in comparison to Isoniazid taken as standard against Mycobacterium tuberculosis. The tested compound 4d exhibited strong antitubercular activity with MIC value of 10 μg/ml. The chromenyl moiety in these compounds is substituted with electron withdrawing groups. However, methyl-substituted chromenyl phosphonate 4e displayed weak antitubercular activity as revealed by the diameter of its inhibition zone (MIC; 120 μg/ml). Thus electron withdrawing groups particularly nitro group reinforced the antitubercular activity of synthesized chromenylphoshonates 4a-e. The reason behind the observed activity trend seems to be the way in which the molecules bind at the target active site.
| Compound code | clogP | H-bond donors | H-bond acceptors | Molecular weight | Zone of inhibition (mm)* |
Minimum inhibitory concentration (µg/ml) |
|---|---|---|---|---|---|---|
| 4a | 2.969 | 1 | 4 | 342.0136 | 19.11 ± 0.21 | 20 |
| 4b | 3.119 | 1 | 4 | 385.9831 | 17.23 ± 0.89 | 20 |
| 4c | 2.116 | 1 | 4 | 308.0726 | 13.49 ± 0.65 | 60 |
| 4d | 2.159 | 1 | 5 | 353.0577 | 24.19 ± 0.99 | 10 |
| 4e | 2.615 | 1 | 4 | 321.9761 | 10.43 ± 0.15 | 120 |
| Isoniazid | -0.69 | 2 | 2 | 137.14 | 25.70 ± 0.63 | 10 |
*Values are given as mean standard deviation (n=3)
Table 3: Antitubercular activity of the synthesized compounds 4a-e
DNA-binding study
UV-Visible spectroscopy
In vitro antitubercular assay revealed that the synthesized molecules inhibited growth and multiplication of M. tuberculosis H37 Rv. Amongst various microbial targets DNA, as an established target for many anti-microbial drugs, was selected as a target for the synthesized molecule 4d. UV-Visible absorption spectroscopy is a commonly used instrumental technique to probe drug-DNA binding [24]. The change in absorption intensity and λmax due to conformational and structural changes in DNA upon interaction with inhibitor characterizes the type of interaction. The magnitude of these changes determines the strength of interaction. Intercalative binding results in hypochromism with a red shift, which is believed to happen due to interaction between the pi anti-bonding orbital (π*) of the intercalated molecule and the pi bonding orbital (π) of DNA base pairs. Electrostatic interaction produces hyperchromism for both DNA and the interacting molecule, which reflects conformational change. Groove binding causes hyperchromism with no or very little red shift, which is believed to be due to interaction between electronic states of interacting molecules and nitrogenous bases of DNA [42]. The UV–visible spectra of 4d with increasing concentrations of sm-DNA is depicted in Figure 2(a). As is evident from the spectra, with increasing concentration of sm-DNA, the absorbance of 4d at 228.8 nm increases (Hyperchromism) with no red shift. This result indicates that 4d interacts with DNA and the possible mode of interaction could be groove binding. The variations obtained in absorbance are useful in quantitative assertion of intrinsic binding constant (Κ) according to Benesi- Hildebrand equation (Eqn. 1).
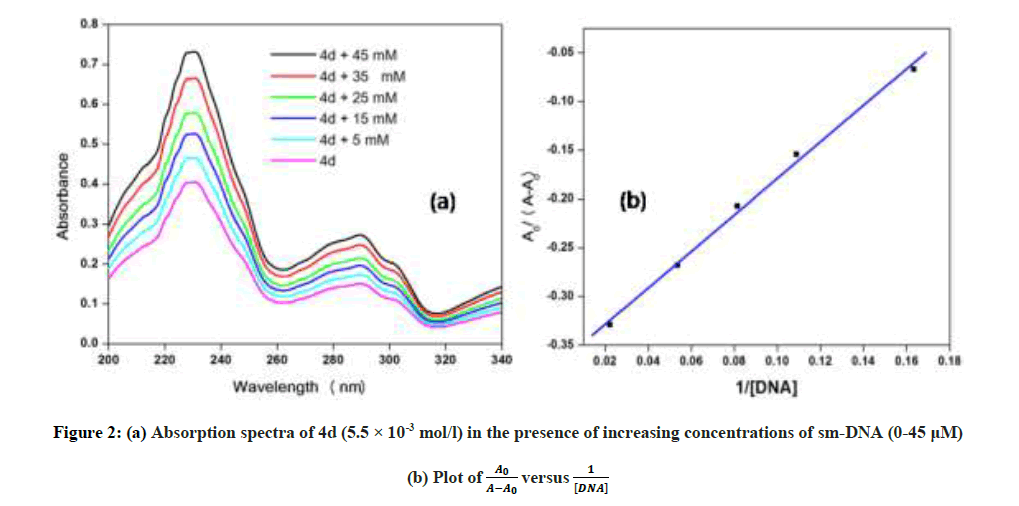
Figure 2: (a) Absorption spectra of 4d (5.5 × 10-3 mol/l) in the presence of increasing concentrations of sm-DNA (0-45 μM)
(b) Plot of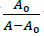 versus
versus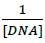
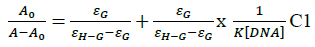
Where, K is binding constant, A0 and A are the absorbance of compound 4d in absence and presence of sm-DNA respectively [43].ε G and ε H-G are absorption coefficients of compound 4d and compound 4d -DNA adduct respectively. The binding constant is evaluated from the intercept to slope ratio of versus plot and was calculated to be 5.317 × 105 M-1 (Figure 2b).
To further distinguish between the mode of interaction, fluorescence quenching titrations and cyclic voltammetric studies were carried out with compound 4d.
Fluorescence spectroscopy
Fluorescence emission spectroscopy was used to probe the binding mode in drug-DNA interactions [42]. Quenching of fluorescence provided valuable information about the interaction of quencher and fluorophore. The fluorescence emission spectrum of 4d showed emission maxima at 479 nm when excited with 240 nm light. As shown in Figure 3a, subsequent addition of sm-DNA to 4d results in quenching of fluorescence with no change in emission maxima. This finding shows that 4d binds with DNA and rules out the possibility of intercalative binding. This result also suggests that 4d interacts with DNA through groove binding. The Stern-Volmer equation (Eqn. 2) was used to study quenching process and to calculate quenching constant:
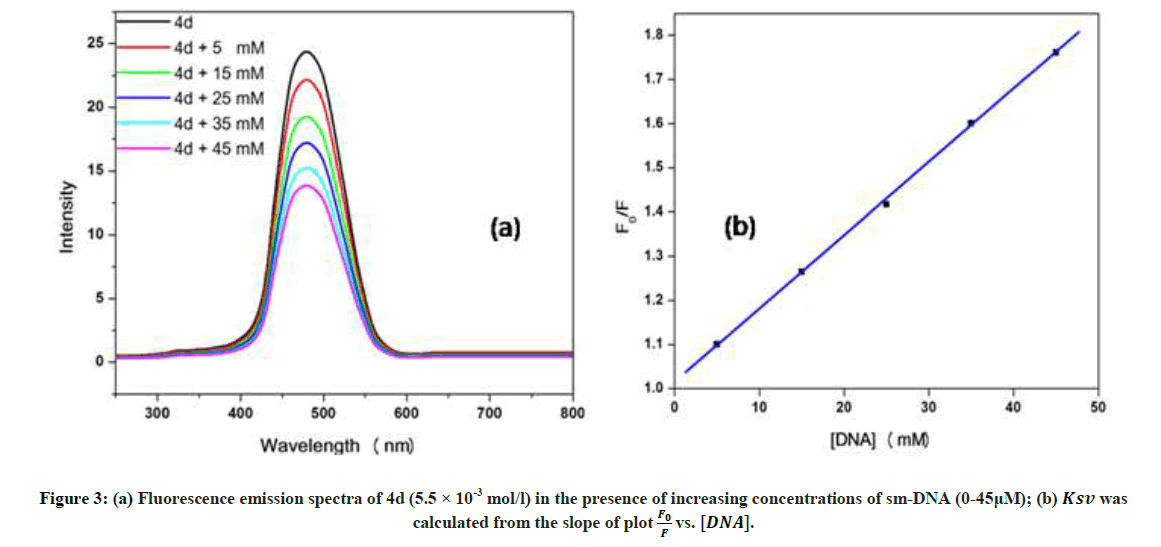
Figure 3: (a) Fluorescence emission spectra of 4d (5.5 × 10-3 mol/l) in the presence of increasing concentrations of sm-DNA (0-45μM);
(b) Ksv was calculated from the slope of plot 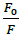 vs. [DNA].
vs. [DNA].
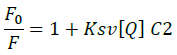
Where, F0 and F-represent fluorescence intensities of 4d in the absence and presence of sm-DNA respectively. Κsv represents the Stern–Volmer constant and [Q] denotes the concentration of quencher (compound 4d). As is evident from Figure 3(b), plot of F0/F (fluorescence quenching) versus concentration of sm-DNA shows a linear relationship. The Stern-Volmer quenching constant (Κsv) of 1.65 × 104 l.m-1 was derived from the ratio of the slope to the intercept of linear Stern-Volmer plot between versus [Q]. This evidence shows that compound 4d interacts with sm-DNA via groove binding.
Cyclic voltammetry
The electrochemical measurements of the compound 4d were carried out by cyclic voltammetry at Glassy carbon Electrode (GCE). The compound 4d exhibited a well-defined, irreversible anodic oxidation peak at -0.940 V (Figure 4a). Cyclic voltammetric measurements were utilized to investigate the interaction of the compound 4d with sm-DNA. As shown in Figure 4a the peak current decreased with increasing concentrations of DNA and a negative shift of peak potential indicating electrostatic interactions of DNA with 4d [44]. The relationship of peak current with DNA concentrations can be employed for the determination of binding constant according to equation (Eqn. 3).
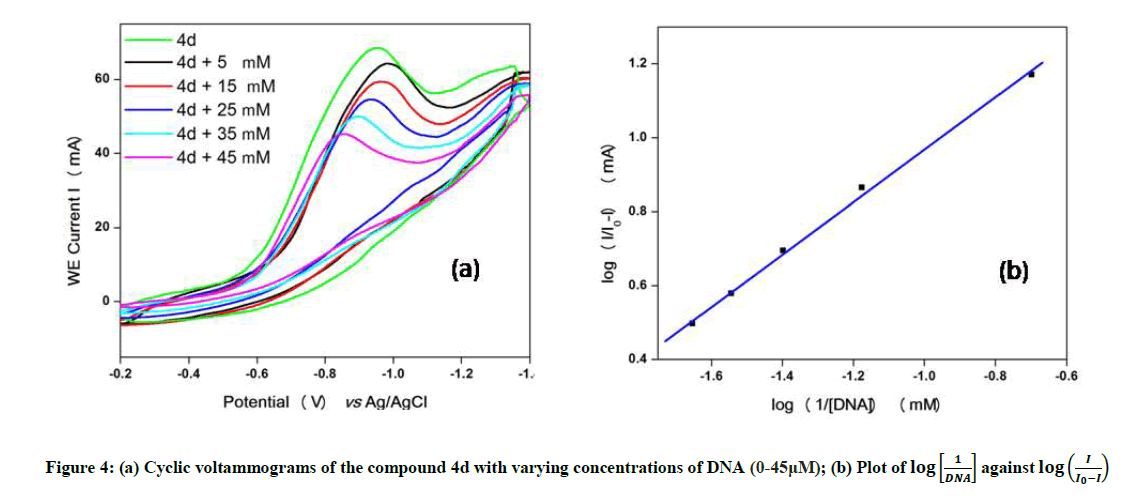
Figure 4: (a) Cyclic voltammograms of the compound 4d with varying concentrations of DNA (0-45μM);
(b) Plot of 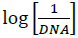 against
against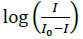
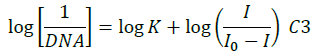
Where, K is binding constant, I and I0 are the peak currents of the drug in absence and presence of DNA respectively [45]. The binding constant K is obtained from the intercept of plot of versus (Figure 4b). The plot of versus was a straight line and the binding constant K was calculated to be 5.67 × 105 M-1. Furthermore, peak to peak separation becomes larger with ΔEP1 and ΔEP2 having the respective values of -0.6 and -0.5 V, unambiguously supported the groove binding mode by the probe 4d with DNA.
Molecular docking studies
Amongst the various target sites for antimicrobial drugs, DNA remains an effective target for bactericidal antibiotics. Drugs interact with DNA by three ways viz., groove binding, intercalation or electrostatic interactions [42]. In the present study, aim of carrying molecular docking was to validate the binding mode of the synthesized compounds with DNA. Molecular docking technique is able to distinguish between groove binding and intercalation [46,47]. As is evident from Figure 5 the docked molecules bind at minor groove of DNA mostly by H-bonding, π-π and hydrophobic interactions. The common nucleotides surrounding the molecules are, DG4, DA5, DA6, DA17, DA18, DT19 and DG22. The highest scored molecule 4d, forms H-bonds of considerable strength with DA5 (3.04Å), DG4 (3.01Å), DG22 (3.05Å) (Figure 6). The formation of multiple H-bonds is a clear indication of strong ligand-DNA complex. Docking scores in the range of -7.2 to -6.0 kcal/mol were predicted for the docked molecules which corroborates high affinity of these molecules towards target DNA (Table 4).
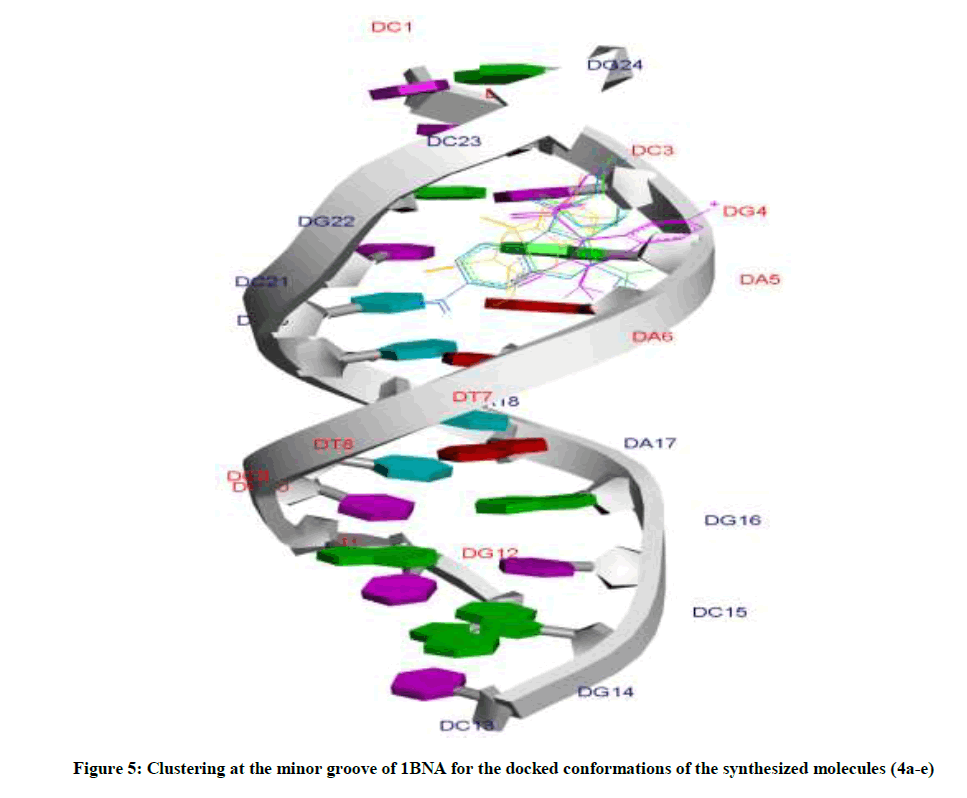
Figure 5: Clustering at the minor groove of 1BNA for the docked conformations of the synthesized molecules (4a-e)
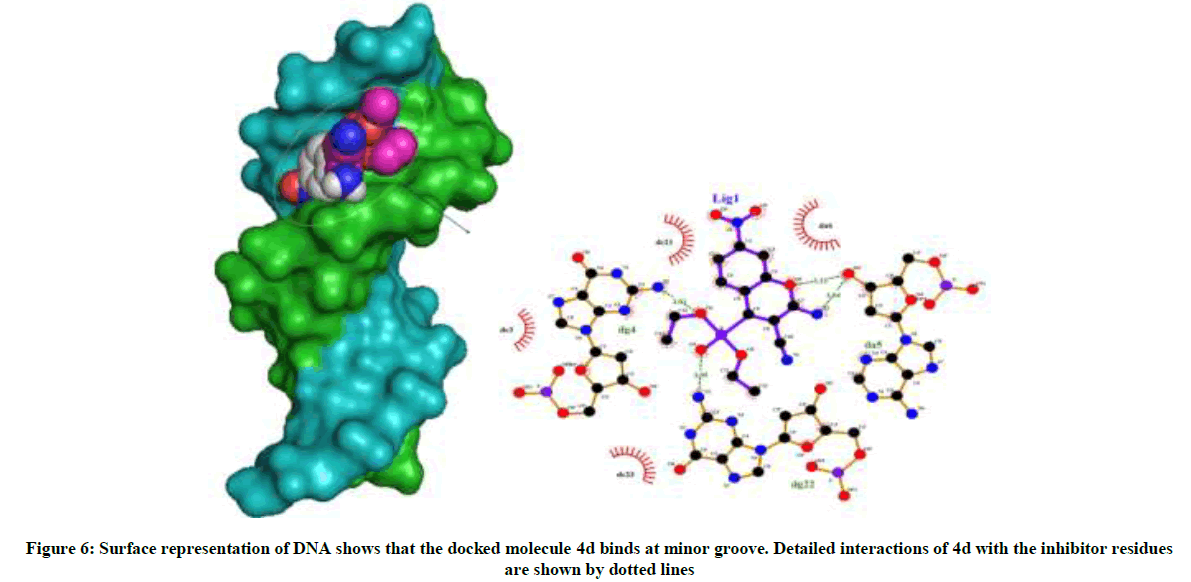
Figure 6: Surface representation of DNA shows that the docked molecule 4d binds at minor groove. Detailed interactions of 4d with the inhibitor residues are shown by dotted lines
| Compound code | Docking score | H-bonding bases with H-bond distance | Other interacting bases |
|---|---|---|---|
| 4a | -6.2 | DA5 (3.18 Å), DG4 (3.07 Å), DG22 (2.97 Å) | DG4, DA5, DG22 |
| 4b | -6.2 | DA5 (3.12 Å), DG22 (2.92 Å) | DA5, DA6, DG22 |
| 4c | -7.2 | DA5 (3.04 Å), DG4 (3.01 Å), DG22 (3.05 Å) | DG4, DA5, DA6, DG22 |
| 4d | -6.1 | DG22 (2.94 Å) | DG22, DA5 |
| 4e | -6.0 | DA18(3.21 Å), DT19(2.80 Å), DA6(2.83 Å) | DA5, DA6, DA17, DA18, DT19 |
Table 4: Docking scores for the compounds 4a-e after docking with Geistenger charges. Interacting and H-bonding residues are shown for each
The most potent molecule 4d was docked with six different fragments of DNA which differed in A/T base pairs. As is evident from Figure 7, the compound 44-Dimethylaminopyridined binds at the minor groove of DNA without exception in all the docked DNA fragments. Crystal structure 1G3X co-crystallized with acridine-peptide drug and 1Z3F were employed to probe intercalative binding mode of 4d as both these structures possess intercalation space. The grid dimensions were set to include the intercalation space. Both co-crystallized acridine–peptide drug and compound 4d were docked at intercalative binding site of 1G3X. The co-crystallized drug binds at the intercalative space in the same conformation as present in X-ray crystal structure (Figure 1). However the compound 4d binds only at the minor groove site both in 1G3X and 1Z3F, it is worth to mention that no π-π stacking interactions were observed. With other DNA fragments the grid dimensions were set to include all the base pairs of DNA for docking, even then the compound 4d specifically binds at the minor groove. Second interesting observation which is evident from Table 5 is that; as the A-T base pair content increases in the DNA fragments, the binding affinity of 4d towards DNA increases. This shows preferential binding affinity of 4d towards A-T base pairs. It is evident from Table 5 that the docked molecule 4d interacts with DNA through non-covalent interactions, H-bonding and van der Waals interactions. The formation of multiple H-bonds is clear evidence in favour of strong ligand-DNA interaction. Docking scores in the range of -7.4 to -6.2 kcal/mol were predicted for 4d when docked with different fragments of DNA. From these results we conclude that the synthesized molecules have potential to bind at the minor groove of DNA and could exert antitubercular as well as anticancer effect by interfering with the normal functioning of DNA.
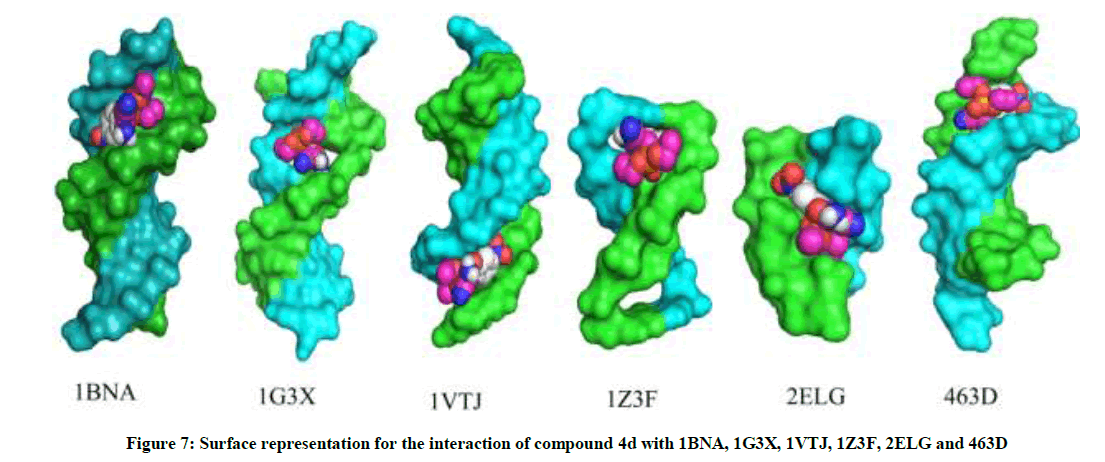
Figure 7: Surface representation for the interaction of compound 4d with 1BNA, 1G3X, 1VTJ, 1Z3F, 2ELG and 463D
| PDB ID | Base pair sequence | ΔG (kcal/mol) |
H-bonding bases | No of H-bonds |
Grid dimensions |
|---|---|---|---|---|---|
| 2ELG | (CGCGCG)2 | -6.2 | ˗ | 00 | 4.917, 0.628, 5.881 |
| 1Z3F | (CGATCG)2 | -6.5 | DG2(3.19 Å), DG6(3.0 Å) | 02 | 0.861, 17.332, 37.318 |
| 463D | (CGCGAATTCGCG)2 | -6.3 | ˗ | 00 | -7.257, 13.496, 1.221 |
| 1VTJ | (CGCGATATCGCG)2 | -6.5 | DA17 (3.11 Å), DG10 (3.23 Å), DC9 (3.43 Å), DC11 (3.35 Å) | 04 | 14.278, 22.413, 75.817 |
| 1BNA | (CGCGAATTCGCG)2 | -7.2 | DA5 (3.04 Å), DG4 (3.01 Å), DG22 (3.05 Å) | 03 | 12.833, 21.133, 8.694 |
| 1G3X | (CGCGAATTCGCG)2 | -7.4 | DA606 (3.04 Å) | 01 | 59.860, 52.124, 59.663 |
Table 5: DNA sequences for DNA fragments with binding affinity of compound 4d with these fragments is in units of kcal/mol. H-bond forming bases/residues are shown
Conclusion
We have reported here a facile and environmentally benign synthesis of (2-Amino-3-cyano-4H-chromen-4-yl)-phosphonates from readily available precursors under ultrasonic irradiation. The synthetic strategy is superior in terms of novelty, scope, safe handling and starting material availability. Operational simplicity, mild reaction conditions, reduced reaction times and ease of product isolation are notable features of this protocol. The reaction favours better functional group tolerance and is high-yielding. All the compounds 4a-e showed promising antitubercular activity. Moreover, the present study indicated that the presence and relative position of nitro group (4d) significantly affected the behaviour of chromenyl phosphonates as novel inhibitors of enoyl-ACP reductase. Utilizing UV-Visible, fluorescence, cyclic voltammetry and molecular docking techniques, DNA was probed as a drug target. It was found that the most potent molecule 4d bound at the DNA minor groove and involved van der Waals, H-bonding and hydrophobic interactions. High binding affinity (-7.4 kcal/mol) and binding constant 5.3 × 105 M-1 was calculated for compound 4d. The present work will access a further increase of the diversity within the (2-Amino-3-cyano-4H-chromen-4-yl)- phosphonate family and to predict their possible interaction mode with sm-DNA.
Acknowledgement
G.A. Khan, U.J. Pandit, and M. Sheikh acknowledges UGC, New Delhi for financial support. One of the authors (J.A. War) would like to acknowledge the financial support of DST, New Delhi in the form of INSPIRE fellowship (IF-120399). Authors are also thankful to SIL of Dr. Harisingh Gour Central University, Sagar (M.P), IIT Guwahati and IISER Bhopal, India for providing instrumental facilities. The authors are thankful to Mr. A. Kumar Department of Zoology, Dr. Harisingh Gour Central University, Sagar (M.P) for his help in carrying out biological tests.
References
- M. Basangouda, V.B. Jambagi, N.N. Barigidad, S.S. Laxmeshwar, V. Devaru, Narayanchar, Eur. J. Med. Chem., 2014, 74, 225.
- K. Sayed, P. Bartyzel, X.Y. Shen, T.L. Perry, J.K. Kjawiony, M.T. Hamann, Tetrahedron., 2000, 56, 949.
- A. Nakhi, B. Prasad, U. Reddy, R.M. Rao, S. Sandra, R. Kapavarapu, D. Rambabu, G.R. Krishna, C.M. Reddy, K. Ravada, P. Misra, J. Iqbal, M. Pal, Med. Chem. Commun., 2011, 10, 1006.
- Z.Q. Xu, W.W. Barrow, W.J. Suling, L. Westbrook, E. Barrow, Y.M. Lin, Bioorg. Med. Chem., 2004, 12, 1199.
- G.A. Khan, J.A. War, G.A. Naikoo, U.J. Pandit, R. Das, J. Saud. Chem. Soc., 2016.
- K. Johnnson, D.S. King, P.G. Schultz, J. Am. Chem. Soc., 1995, 117, 5009.
- D.E.C. Corbridge, Biochemistry and Uses, 5th Ed., Elsevier, Amsterdam, 1995.
- M.C. Allen, W. Fuhrer, B. Tuck, R. Wade, J.M. Wood, J. Med. Chem., 1989, 32, 1652.
- P. Kafarski, B. Lejczak, Phosphorus, Sulfur and Silicon and the Related Elements., 1991, 63, 193.
- D. Enders, A.S. Dizier, M.I. Lannou, A. Lenzen, Eur. J. Org. Chem., 2006, 1, 29.
- A.N. Pudovik, I.V. Konovalova, Synthesis., 1979, 81.
- A. Michaelis, R. Kaehne, Berichte der Deutschen Chemischen Gesellschaft., 1898, 1048.
- R.Y. Guo, Z.M. An, L.P. Mo, R.Z. Wang, H.X. Liu, S.X. Wang, Z.H. Zhang, ACS Comb. Sci.,2013, 15, 557.
- B. Rotstein, S. Zaretsky, V. Rai, A.K. Yudin, Chem. Rev., 2014, 114, 8323.
- A. Domling, W. Wang, K. Wang, Chem. Rev., 2012, 112, 3083.
- G.H. Posner, Chem. Rev., 1986, 86, 831.
- G.A. Khan, J.A. War, A. Kumar, I. Sheikh, A. Saxena, R. Das, J. Taibah Univ. Sci., 2016, 11(6), 910-921.
- K.M. Khan, W. Jamil, N. Ambreen, M. Taha, S. Perveen, G. A. Morales, Ultrasonics Sonochem.,2014, 21, 1200.
- R.H. Nia, M. Mamaghani, K. Tabatabaeian, F. Shirini, M. Rassa, Bioorg. Med. Chem. Lett., 2012, 22, 5956.
- J. Safaria, L. Javadian, Ultrasonics Sonochem.,2015, 22, 341.
- J.H. Shi, TT. Liu, M. Jiang, J. Chen, Q. Wang, J. Photochem. Photobiol. B., 2015, 147, 47.
- P.B. Dervan, B.S. Edelson, Curr. Opin. Struct. Biol., 2003,13, 284.
- U.J. Pandit, G.A. Khan, G.A. Naikoo, S. Wankar, K.K. Raj, S.N. Limaye, Der Pharma Chemica., 2017, 9(1), 82.
- J.A. War, S.K. Srivastava, S.D. Srivastava, Luminescence.,2016.
- D.W Lawrence, A.L Barry, R.O. Toole, J.C. Sherris, Appl. Microbiol., 1972, 24, 240.
- O. Trott, A.J. Olson, J. Comp. Chem., 2010, 31, 455.
- H. Ohishi, Y. Tozuka, Z. Da-Yang, T. Ishida, K. Nakatani, Biochem. Biophys. Res. Commun.,2007, 358, 24.
- S.L. Lam, L.N. Ip, J. Biomol. Struct. Dyn., 2002, 19, 907.
- J. Liu, J.A. Subirana, J. Biol. Chem., 1999, 274, 24749.
- N.G. Abrescia, L. Malinina, J.A. Subirana, J. Mol. Biol., 1999, 294, 657.
- H.R. Drew, R.M. Wing, T. Takano, C. Broka, S. Tanaka, K. Itakura, R.E. Dickerson, Proceedings of the National Academy of Sciences., 1981, 78, 2179.
- L. Malinina, M. Soler-López, J. Aymamí, J.A. Subirana, Biochemistry (Mosc)., 2002, 4, 9341.
- J.B. Cross, D.C. Thompson, B.K. Rai, J.C. Baber, K.Y. Fan, Y. Hu, C. Humblet, J. Chem. Inform. Modeling., 2009, 49, 1455.
- M. Frisch, G. Trucks, H.B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, Inc., Wallingford, CT, 2009, 200.
- J.S. Yadav, B.V.S. Reddy, K. Praneeth, Tet. Lett., 2008, 49, 199.
- R. Elayaraja, R.J. Karunakaran, Tetrahedron Lett., 2012, 53, 6901.
- S. Nagarajan, T.N. Das, S. Nagarajan, T.M. Das, Carbohydrate Res., 2009, 344, 1028.
- J.H. Park, Y.R. Lee, S.H. Kim, Tetrahedron.,2013, 30, 1.
- M. Rajasekhar, K.U.M. Rao, C.S. Sundar, N.B. Reddy, S.K. Nayak, C.S. Reddy, Chem. Pharm. Bull.,2012, 60, 854.
- P. Dai, G. Zha, X. Lai, W. Liu, Q. Gan, Y. Shen, RSC Adv.,2014, 4, 63420.
- R.M.N. Kalla, J.S. Choi, J.W. Yoo, S.J. Byeon, M.S. Heo, I.I. Kim, Eur. J. Med. Chem., 2014, 76, 61.
- M. Sirajuddin, S. Ali, A. Badshah, J. Photochem. Photobiol. B., 2013, 124, 1.
- Y. Li, Z.Y. Yang, J. Fluoresc., 2010, 20, 329.
- N. Li, Q. Ma, C. Yang, L. Guo, X. Yang, Biophy. Chem., 2005, 116, 199.
- B. Rafique, A.M. Khalid, K. Akhtar, A. Jabbar, Biosens. Bioelectron., 2013, 44, 21.
- C.G. Ricci, P.A. Netz, J. Chem. Inform. Mod., 2009, 49, 1925.
- M. Al-Rashida, S. Ahsen, RSC Advances., 2015, 5, 72394.