Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 4
A Novel Method for the Estimation of Budesonide in Human Plasma by Using LC-MSMS
Raveendra Babu Gudimitla1, Lakshmana Rao Atmakuri2* and Venkateswara Rao Jangala3
1Department of Pharmaceutical Analysis, A.K.R.G. College of Pharmacy, Nallajerla, Andhra Pradesh, India
2Department of Pharmaceutical Analysis, V.V. Institute of Pharmaceutical Sciences, Gudlavalleru, Andhra Pradesh, India
3Department of Pharmaceutical Analysis, St Pauls College of Pharmacy, Turkayamijal, Hyderabad, Telangana, India
- *Corresponding Author:
- Lakshmana Rao Atmakuri
Department of Pharmaceutical Analysis
V.V. Institute of Pharmaceutical Sciences
Gudlavalleru, Andhra Pradesh, India
Abstract
A novel method for the estimation of Budesonide in human plasma by using LC-MS-MS and the analyte is budesonide and internal standard is levenorgestreol were extracted with the tertbutyl methyl ether: n-hexane (70:30, v/v) from human plasma. The chromatographic severance was attained of the peak using Agilent Zorbax Eclipse XDB-C8, (100 mm × 4.6 mm, 3.5 μm) column with a run time is 2.5 min. Budesonide and levenorgestreol were recorded at the total ion current of their relevant multiple reaction monitoring. The LC-MS-MS system composed an Agilent 1100 infinity combined with an AB Sciex Qtrap® 4000 thermo Finnigan TSQ quantum discovery triple quadruple mass spectrometer. All of the parameters must be validated like selectivity, accuracy, precision, linearity, lower limit of quantification, matrix effect, recovery reached the acceptance criteria under the following of ICH guidelines. Budesonide have checked the various stability studies like short term stability at 25°C, long term stability for 55 days at -70°C, wet extract stability for 54 h, auto sampler stability for 63 h, bench top stability for 14 h and freeze-thaw stability at -60°C. Hence, it can be used for routine drug analysis and bioequivalence studies of budesonide in human plasma samples.
Keywords
Budesonide, Levenorgestreol, Estimation, Human plasma, LC-MS/MS and Validation
Introduction
Budesonide was a glucocortico steroid and its chemical name is 16,17-(butylidenebis(oxy))-11,21-hydroxy-(11-β,16-α)-pregna-1,4-diene-3,20- dione. Budesonide designated for the asthma, nasal polyposis and Crohn’s disease [1] and it was used for long term management of asthma and chronic obstructive pulmonary disease with the help of inhaled corticosteroid therapy [2]. A relevant number of studies for estimation of budesonide have been reported, the methods employed includes UV spectroscopy [3,4] high performance liquid chromatography [5-7] and liquid chromatography-mass spectroscopy [8-10]. Pharmacokinetics and pharmacodynamics of budesonide have been measured in healthy volunteers [11,12] and primary biliary cirrhosis patients [13] However, no studies regarding the estimation of budesonide in human plasma by using Liquid Chromatography-Mass Spectroscopy-Mass Spectroscopy (LC-MS-MS) has published so far. The goal of the present study was estimation of budesonide in human plasma by using LC-MS-MS. Besides, until now, no studies have been reported in scientific literature regarding the data of full bio-analytical validation of estimation of budesonide in human plasma by using LC-MS-MS. This paper reports the simple, sensitive, rapid, precise and accurate method for the estimated of budesonide in human plasma by using LC-MS-MS. Based on the data obtained the LC-MS-MS has been applied for analysis of commercial and bioequivalence studies of budesonide samples.
Materials and Methods
Materials
Budesonide (≥ 99%) was purchased from Aarti Industries Limited (Mumbai, India). Levonargestrel (≥ 98%) was purchased from Clearsynth Labs (Mumbai, India). Acetonitrile (≥ 95.8%) and methanol (≥ 98.5%), tert butyl methyl ether (≥ 99.5%), HPLC grade were purchased from J.T. Baker (Philipsbur, USA). Formic acid, analytical grade, n-hexane and water, both HPLC grade, were purchased from Merck Limited (Mumbai, India).
Instrument
The LC-MS-MS system composed an Agilent 1100 infinity combined with an AB Sciex Qtrap® 4000 thermo Finnigan TSQ quantum discovery triple quadruple mass spectrometer and the control was done by the Analyst software version 1.4.2. Ultrapure water (18 MΩ/cm) was produced using Ultra ClearTm system (Richfield, USA). The pH of the buffer solution was measured using a pH/mv-meter Consort P501 (Turnhout- Belgium), provided with combined pH electrode. UV-Visible spectra were recorded on a Jasco V-530 spectrophotometer (Tokyo, Japan), in 10 mm quartz cells.
Solutions of LC-MS-MS
A stock solution of budesonide containing 200 μg/ml was prepared in 10 ml volumetric flask, by dissolving 0.0020 g of budesonide in methanol (HPLC grade). The flask was filled up to the mark with the same solvent. A stock solution of levenorgestreol (Internal standard) containing 200 μg/ml was prepared in 10 ml volumetric flask, by dissolving 0.0020 g levenorgestreol (Internal standard) in acetonitrile and water, 50:50, v/v) as diluent. The flask was filled up to the mark with same solvent.
Working solutions containing all of the analytes were prepared in the range 0.100 ng/ml to 3.00 ng/ml using (Acetonitrile: Water, 50:50, v/v) as diluting the solvent. All the analytes solutions were prepared daily and kept at 2-8°C before and between injections to prevent sample degradation. An ammonium formate buffer solution pH=4.20 was prepared by mixing of 0.1578 g of ammonium formate in 500 ml of ultrapure water. If necessary he pH could be adjusted to pH=4.20 with 0.1% formic acid solution.
Sample collection and preparation
Blood samples were collected into blank tubes and centrifuged for 5 minutes at approximately 4000 rpm at 4°C. The plasma samples (supernatant) was separated and stored at -20°C until analysis. Budesonide was extracted from the plasma samples where with protein precipitation. Concisely, 200 μl of plasma sample were extracted in 4 ml of extraction solvent (tert-butyl methyl ether: n-hexane, 70:30, v/v) including the internal standard (Levenorgestreol at 200 ng/ml). The mixture was vortexed for 20 min and centrifuged at 4000 rpm, for 5 min at 4°C. 3 ml supernatant layer was separated and evaporated under gentle stream of nitrogen gas at 45°C upto dryness. Then the residue was reconstituted with 500 μl mobile phase (Acetonitrile: 5 Mm ammonium formate buffer in 0.1% formic acid, 60:40, v/v) and loaded into a pre labelled auto sampler vials.
Chromatographic and mass spectrometric conditions
Chromatographic studies were performed using an Agilent Zorabax Eclipase XDB-C8 purchsed from Thermo Electron Corporation, USA, with the dimensions 150 mm × 4.6 mm and 3.5 μm particle size and the injection volume was 5 μl. The mass spectrometer was operated under positive ionization mode and the detection of the analytes was based on multiple reaction monitoring of m/z 431.3/323.1 and 313.40/245.30 for budesonide and levenorgestreol, respectively. The ion spray voltage, the source temperature, declustering potential, collision cell exit potential, entrance potential and dwell time were sets at 5000V, 500°C, 90V, 11V, 10V and 200 ms, respectively.
Validation of the method
The method was validated according to the ICH guidelines [14].
Software
Calculating the molecular mass of analyte and internal standard by using version 1.4.2 for Analyst software.
Results and Discussion
Method development
Budesonide and all of the compounds enclosed in this study ionize according to the pH the mobile phase. The development of the new LC-MS/ MS method is based on the behaviour of ionisable compounds that have the retention closely related to the pH of the mobile phase. The results shown in Figures 1 and 2.
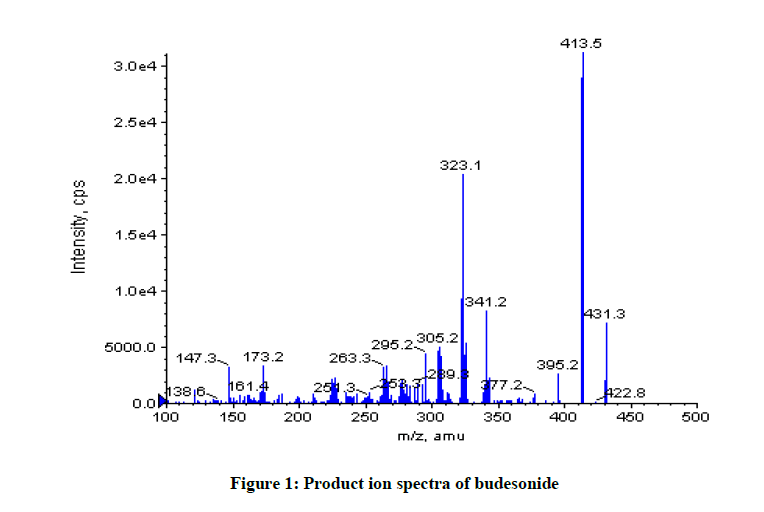
Figure 1: Product ion spectra of budesonide
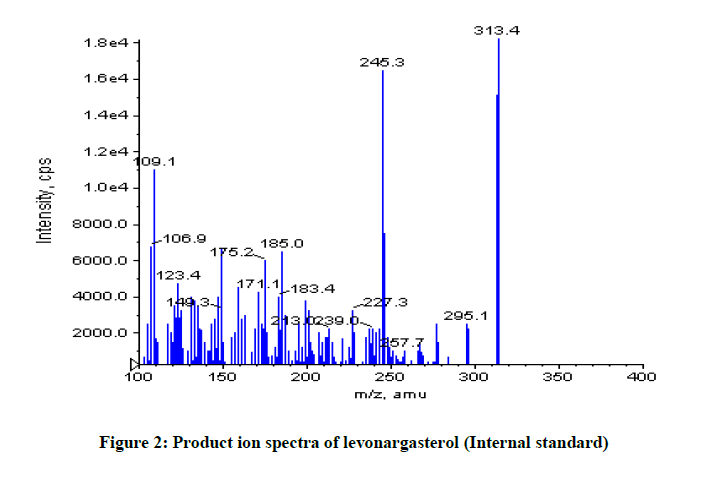
Figure 2: Product ion spectra of levonargasterol (Internal standard)
As shown Figure 1, budesonide exists molecular mass: Parent peak molecular mass is 413.5 m/z and doughtier peak molecular mass is 323.1 m/z, respectively. As shown Figure 2, levonargasterol exists molecular mass: Parent peak is 313.4 m/z and doughtier peak molecular mass is 245.3 m/z, respectively.
Method validation
The final developed method (As described in the experimental section) was validated, checking for linearity, precision, accuracy, matrix effect and stability studies.
Linearity
Linearity has been checked directly for each compound studied in this paper by dilution of a standard stock solution. The slop and y-intercept were provided as equation to gather with the correlation coefficient in order to demonstrate the linearity of the method. Linearity was established for working solution containing concentration between 0.103-3.010 ng/ml for Budesonide. The equation was obtained y=0.101+0.00158 (r2=0.9989) shown in Figure 3.
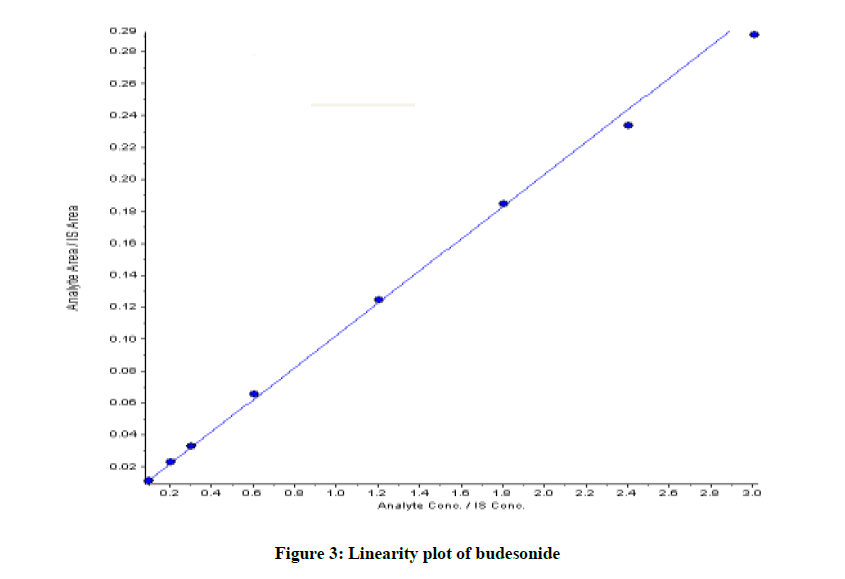
Figure 3: Linearity plot of budesonide
Precision
The precision was estimated by both with-in batch precision and between batch precision. According to the international rules of validation of with-in batch precision was checked on four different concentrations like Higher Quality Control (HQC), Medium Quality Control (MQC), Lower Quality Control (LQC) and Lower Limit of Quality (LLOQ) Quality Control (QC).
Between batches precision was established on another day, by a different investigator using percent coefficient of variation. The obtained values for %CV are below 15% (of HQC, MQC and LQC) and below 20% (LLOQ QC) showing a good precision of the method. The results are present in Table 1.
| Within-batch precision (n=6) for budesonide | |||
|---|---|---|---|
| Sample ID | Concentration found (mean ± SD, n=6; µg/ml) | Precision (%) | Accuracy (%) |
| HQC | 2.633 ± 0.155 | 5.26 | 105.58 |
| MQC | 1.570 ± 0.073 | 4.65 | 99.61 |
| LQC | 0.268 ± 0.010 | 3.86 | 103.4 |
| LLOQ QC | 0.114 ± 0.010 | 9.19 | 107.17 |
| Between batch precision for budesonide | |||
| Sample ID | Concentration found (mean ± SD, n=6; µg/ml) | Precision (%) | Accuracy (%) |
| HQC | 2.843 ± 1.94 | 6.85 | 107.98 |
| MQC | 0.543 ± 0.033 | 6.26 | 104.47 |
| LQC | 0.266 ± 0.017 | 6.73 | 102.33 |
| LLOQ QC | 0.113 ± 0.008 | 7.81 | 105.86 |
Note: S.D.=Standard deviation; %CV=Percent coefficient of variation
Table 1: Precision data of budesonide
Accuracy
Accuracy was determined by calculating the recovery of each analyte at three different concentration level like HQC, MQC and LQC. The obtained values for the recoveries were in the range of 72.48-81.48%. The results are present in Table 2.
| Parameter | LQC | MQC | HQC |
|---|---|---|---|
| % of Recovery | 76.43 | 72.48 | 81.48 |
| % C.V | 2.92 | 6.71 | 4.54 |
Table 2: Accuracy data of budesonide
Matrix effect
It is combined effect of all components of the sample other than the analyte. The matrix effect checked by three different concentration levels, these are HQC, MQC and LQC. The %CV values below 15% showing good matrix effect of the method. The results are presented in Table 3.
| Parameter | HQC | MQC |
|---|---|---|
| %Accuracy | 100.12 | 99.89 |
| S.D (+/-) | 0.002 | 0.065 |
| %CV | 0.95 | 2.50 |
Table 3: Matrix effect data of budesonide
Stability studies
Stabilities were estimated like at low and high concentrations of sample of budesonide in plasma. Checking the stabilities like short term stability at -20°C, long term stability for 55 days at -70°C, wet extract stability for 54 h, auto sampler stability for 63 h, bench top stability for 14 h and freeze and thaw stability (3) cycles at -70°C, respectively. The %CV values below 15% showing good stability effect of the method. The results are shown in Table 4.
| Parameter | Results | |
|---|---|---|
| HQC | LQC | |
| Short term stability (-20°C) | 105.16% | 103.27% |
| Long term stability (-70°C) 55 days | 105.55% | 105.38% |
| Wet extract stability (54 h) | 104.51% | 99.87% |
| Auto sampler stability (63 h) | 99.00% | 98.21% |
| Bench top stability (14 h) | 93.56% | 95.71% |
| Freeze & thaw stability (3) cycles at -60°C | 107.80% | 100.32% |
Table 4: Stability studies data of budesonide
Conclusion
The proposed LC-MS/MS method was simple, rapid, precise and accurate for determination of budesonide in human plasma. Sample preparation showed high recovery for the quantitative estimation of budesonide in human plasma and this method to allow higher sample throughout put due to the short chromatography time (3.5 min) and simple sample preparation. Thus, the developed LC-MS/MS method was can apply for the bioequivalence and pharmacokinetic studies of budesonide in human plasma samples.
Acknowledgements
The authors are thankful to A.K.R.G. College of Pharmacy, Nallajerla, India for providing necessary facilities for carryout the research work.
References
- K.D. Tripathi, Essentials of Medical Pharmacology, 4th Edi., Medical Publishers, New Delhi, 2001, 230-231.
- C. Christensson, A. Thoren, B. Lindberg, Drug. Saf., 2008, 31, 965-988.
- D.D. Sanap, A.M. Sisodia, S.H. Patil, IJPSR., 2011, 2, 2419-2423.
- G.M. Mallikarjuna, S.A. Ramakrishna, S.M. Shantakumar, J. Appl. Pharm. Sci.,2011, 1, 158-161.
- N.S. Mohanad, D. Yusrida, K.K. Peh, Afr. J. Pharm. Pharmacol., 2010, 4, 878-884.
- R.K. Nanasaheb, P.P. Ashok, A.M. Javed, WJPR., 2014, 3, 1387-1399.
- D. Subhash, S. Sanjay, B. Anil, Ame. J. PharmTech. Res.,2013, 3, 940-947.
- K. Deventer, P. Mikulcikova, H.H. Van, J. Pharm. Biomed. Anal., 2006, 11, 474-479.
- P. Deng, X.T. Duan, X.Y. Chen, Acta Pharmaceutica Sinica., 2008, 43, 76-80.
- A. Szeita, J. Manji, K.W. Riggs, J. Pharm. Biomed. Anal., 2014, 90, 198-206.
- K. Dilger, J. Haiter, H. Bertz, Biol. Blood Marrow Transplant., 2009, 15, 336-343.
- L.C. Karine, A.W. Peter, M. Karen, British. J. Clinic. Pharmacol., 2011, 71, 504-513.
- W. Hempfling, F. Grunhage, K. Hepatology., 2003, 38, 196-202.
- V.S. Lalit, N.P. Bhagwat, V.U. Sharad, Pharmaceutica. Analytica Acta., 2014, 5, 4172.



