Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 6
A Novel RP-HPLC Method for the Simultaneous Assessment of Olmesartan, Amlodipine and Hydrochlorothiazide and its Application to In-vitro Dissolution
Ragaa El Sheikh1, Abdelaziz M. Annadi1 and Ashraf A. Mohamed2*
1Chemistry Department, Faculty of Science, Zagazig University, Zagazig, 44519, Egypt
2Chemistry Department, Faculty of Science, Ain Shams University, Abbassia, Cairo-11566, Egypt
- *Corresponding Author:
- Ashraf A. Mohamed
Chemistry Department
Faculty of Science
Ain Shams University
Abbassia, Cairo-11566, Egypt
Abstract
The development and validation of a simple, rapid, precise and accurate RP-HPLC has been described for the simultaneous assessment and invitro dissolution of Olmesartan (OLM), Amlodipine (AML) and Hydrochlorothiazide (HCTZ) in their combined dosage forms. Separation was performed on Inertsil ODS-3 column (C18, 150 mm x 4.6 mm, 5 μm) using photodiode array (PDA) detection at 255 nm. The mobile phase consisted of phosphate buffer (pH 3.5) and acetonitrile, in a gradient program (time in minutes/acetonitrile %: 0.00/30, 1.00/30, 8.30/70 and 9.00/30) at a 1.3 ml min-1 flow rate. Analytes were perfectly resolved with retention times of 2.59, 4.99 and 7.48 min for OLM, AML, and HCTZ, respectively. Following the recommended procedure, linear calibration graphs extended for 0.20-28.00, 0.05-7.00 and 0.12-17.50 μg ml-1 with detection limits of 0.021, 0.002 and 0.012 μg m;-1 and quantitation limits of 0.063, 0.036 and 0.007 μg ml-1 for the assay and in-vitro drug release of OLM, AML and HCTZ, respectively. In-Vitro dissolution revealed that >96% of the labeled OLM, AML and HCTZ were released from their combined tablets within 20 min. The developed method was validated following the ICH guidelines regarding the system suitability, specificity, linearity, accuracy, precision, and robustness.
Keywords
Olmesartan, Amlodipine, Hydrochlorothiazide, In-vitro dissolution, RP-HPLC, Validation.
Introduction
Angiotensin antagonists are the first line treatment of hypertension. Angiotensin II receptor blockers/antagonists (ARA II) are effective renin– angiotensin modulators that act by displacing angiotensin II from the system. ARA II is used in the treatment of hypertension, diabetic nephropathy and congestive heart failure. They block the activation of AT1 receptors, preventing the binding of angiotensin II. It is now well established that monotherapy is insufficient to control blood pressure disorders in the majority of patients; therefore, most patients will require polytherapy [1-3]. A fixed dose ternary therapy containing 20/5/12.5 mg, Olmesartan (OLM), Amlodipine (AML) and Hydrochlorothiazide (HCTZ), has been recognized of its efficiency and safety in a number of clinical trials [1-3]. The structural formulae of OLM, AML and HCTZ of this effective ternary therapy are given in scheme 1 [1-3].
Scheme 1: structural formulae of OLM, AML and HCTZ
Analytical methods have been described for the determination of binary and ternary mixtures of OLM, AML and HCTZ included spectrophotometric [3-5], HPTLC [6-9], HPLC [10-17], and UPLC [18-21]. Among these, one method described the in-vitro dissolution studies of the binary (OLM, AML) [11]; however, four methods described the ternary (OLM, AML, HCTZ) combinations [14-17]. However, the high detection limits and poor sensitivity are major disadvantages [3-7,9-11,13-21].
The aim of this work is to develop a highly sensitive RP-HPLC method for the rapid and simultaneous assessment of assay content and in-vitro dissolution release of HCTZ, AML and OLM in their ternary therapy dosage forms and to validate the proposed method according to the International Conference on Harmonization (ICH) guidelines [22].
Experimental
Instrumentation
A Waters HPLC system (Waters, Milford, UK) model 2690 equipped with an (C18, 150 x 4.6 mm, 5 μm) Inertsil ODS-3 column (Tokyo, Japan) and a photo diode array (PDA) detection was used. A calibrated Mettler Toledo pH meter model S20 (Columbus OH, USA) and a TDT-08L Electrolab Dissolution tester (Mumbai, Maharashtra, India) were also used. A Barnstead Nanopure infinity ultrapure water system (Dubuque, IA, USA) was used to daily provide ASTM grade I ultrapure water. Millipore Polyvinylidene Fluoride (PVDF) syringe filters with 0.22 μm pore size and 33 mm diameter were used for filtration.
Materials and reagents
United States Pharmacopeial (USP) grade Olmesartan medoxomil, Amlodipine besylate, and Hydrochlorothiazide were purchased from Sigma (St. Louis, MO, USA). Reagents of ACS grade of sodium dihydrogen phosphate, potasium monohydrogen phosphate, phosphoric acid, and HPLC grade acetonitrile and methanol were purchased from Merck (Darmstadt, Germany) or Scharlau (Barcelona, Spain). An elution buffer of pH 3.50 was prepared by dissolving 3.90 g sodium dihydrogen phosphate dihydrate in water, adjusting the pH to the desired value with phosphoric acid and diluting to 1 l in a volumetric flask. In addition, a phosphate buffer dissolution medium was prepared, following the Food and Drug Administration (FDA) guidance [23], by dissolving 20.40 g K2HPO4 and 21.18 g NaH2PO4 in 6 l of water and adjusting the pH to 6.80 (± 0.05) with sodium hydroxide or phosphoric acid solutions.
Chromatographic conditions
In the optimized RP-HPLC procedure, chromatographic separation was conducted on a Waters 2690-HPLC system with an Inertsil ODS-3 column and a detection at 255 nm. The mobile phase consisted of the phosphate buffer of pH 3.5 and acetonitrile, in a gradient program (time in minutes/acetonitrile % : 0.00/30, 1.00/30, 8.30/70 and 9.00/30) at a flow rate of 1.3 ml min-1. The column temperature was 30°C and the injection volume was 50 μl.
Procedures
Solutions of standards and samples
A stock mixed solution was prepared by dissolving 25.0 mg of HCTZ, 10.0 mg AML and 40.0 mg OLM in 100 ml of methanol in an ultrasonic bath. Appropriate aliquot volumes of this solution were diluted with the eluent (30% acetonitrile/phosphate buffer of pH 3.50), to prepare various working solutions over the concentration ranges of (0.12-17.50), (0.05-7.00) and (0.20-28.00) μg ml-1 HCTZ, AML and OLM, respectively, for the assay and in-vitro drug release profile investigations. However, for sample preparation, ten tablets of the test sample were grinded in a mortar. Appropriate fine powder portion was exactly weighed, dissolved in a 50 ml methanol plus 50 ml of the phosphate buffer dissolution medium of pH 6.80, in the ultrasonic bath, filtered using 0.20 μm membrane filter, quantitatively transferred and diluted to 100 ml in a volumetric flask. Appropriate volumes of this test solution were diluted with the eluent (30% acetonitrile/phosphate buffer of pH 3.50, in 10 ml volumetric flasks.
In-vitro dissolution
Dissolution studies on the fixed-dose combined tablet formulation (25 mg HCTZ, 10 mg AML and 40 mg OLM) were performed using USP Apparatus II (paddle method) with six replicates at 37 ± 0.5°C. A 900 ml of the phosphate buffer, dissolution medium of pH 6.80, was used and the paddle rotation speed was kept at 50 rpm [23]. At predetermined intervals of 5, 10, 15, 20, 30 and 45 min; a 1 ml aliquot was withdrawn and replaced with a similar volume of fresh medium to keep a constant total volume. This aliquot was filtered using 0.20 μm membrane filter, and a 500 μl portion was mixed with 500 μl of the mobile phase. The concentrations of OLM, AML and HCTZ in these aliquots were determined simultaneously by the developed RP-HPLC method.
Method validation
The developed RP-HPLC method for the simultaneous assessment of OLM, AML and HCTZ was systematically validated following the International Conference on Harmonization (ICH, Q2, R1) guideline validation for analytical procedure, regarding system suitability, linearity, accuracy, precision, specificity, and robustness [22].
The suitability of the proposed RP-HPLC method was evaluated by calculating the number of theoretical plates of higher than 2000, tailing factor < 2.0 and RSD% ≤ 2.0, for five replicate injections [24]. The proposed RP-HPLC method specificity was determined by attesting the interferences of blank/placebo, impurity peaks at the respective retention times of OLM, AML and HCTZ, respectively. Standard and sample solutions were prepared as described above. Placebo solution was similarly prepared by taking a placebo and omitting the three analytes. Impurities were prepared at a 5% level with respect to the analytes concentrations. Solutions were injected and the obtained chromatograms were verified for the respective peaks purity as well as interferences of the blank, placebo and impurity peaks.
The analytical procedure’s linearity is its ability to register test results that are linearly proportional to the analyte concentration. The proposed method’s linearity was determined by injecting three replicates of seven concentration levels covering the concentration ranges of (0.20-28.00), (0.05-7.00) and (0.12-17.50) μg ml-1 OLM, AML and HCTZ, respectively. Linear regression’s analysis was used for data evaluation in terms of the regression coefficient (r2 > 0.999), y-intercept and slope of regression lines obtained by plotting peak areas against concentrations of standards. The range was verified by testing at the proposed minimum 50% and maximum 150% levels relative to the sample concentration. In addition, the LOD and LOQ were calculated as 3.3 σ/S and 10 σ/S, respectively; where σ and S was the standard deviation of the y-intercept and the slope of the calibration line, respectively.
The accuracy is the closeness of test result to the true value. Accuracy was determined by means of recovery experiments, by spiking of active drug to placebo formulations and analysis following the recommended procedure. The accuracy was calculated from the test results as the percentage of the analyte recovered by the assay [22].
The precision reflects the level of agreement between replicate measurements and can be expressed as a relative standard deviation (% RSD). The precision of the method was attested by six replicate injections for each of three concentration levels of the mixed standard solutions containing 50%, 100% and 150% levels of sample concentration. Repeatability (Within-run) and reproducibility (between-run) were assessed.
Results and Discussion
Preliminary investigations
The RP-HPLC separation and simultaneous assessment of OLM, AML and HCTZ in their ternary therapy was adopted in this work due to its simplicity, selectivity and suitability. Optimization of experimental parameters is generally performed to achieve some important criteria including high sensitivity, specificity, accuracy, precision, and speed of analysis.
Photo diode array spectra were recorded (200-400 nm) for OLM, AML and HCTZ and the registered wavelength maxima were, in agreement with the USP data [23], at 249, 237 and 275 nm, respectively. Therefore, a wavelength of 255 nm was adopted for assessment to achieve a good response, towards the three investigated drugs in their ternary mixture, while being away from any interference that may exist at lower wavelengths.
One of the main targets in developing the current RP-HPLC method was to achieve a good resolution between the three drugs and any possible interference from the dissolution medium, placebo or eluent. In this regard, various mobile phases and compositions were tested to obtain a good resolution and sharp symmetric peaks for OLM, AML and HCTZ. This target was achieved only with the phosphate buffer-acetonitrile eluent. Phosphate buffer alone and phosphate buffer–methanol combinations gave low resolution and low sensitivity compared to phosphate buffer– acetonitrile eluent compositions. Scouting of buffer pH (3.00-4.00) and % acteonitrile (10-80%) has been performed. Sharp, symmetric and well resolved peaks were obtained at buffer pH=3.50 ± 0.05, and % acetonitrile of 30 ± 2% at a flow rate of 1.0 ml min-1. However, a relatively long run-time of about 20 min was observed.
Therefore, to improve the method performance and decrease the run time, we tested a gradient program where best performance was exhibited with (time in minutes/acetonitrile %: 0.00/30, 1.00/30, 8.30/70 and 9.00/30) at a flow rate of 1.3 ml min-1. Under these conditions, HCTZ, AML, and OLM were excellently resolved with retention times of 2.59, 4.99 and 7.48 min, respectively, as shown in Figure 1.

Figure 1: Chromatogram of HCTZ, AML and OLM. Conditions were those of the recommended procedure
Selectivity and peak purity
There was no peak of placebo, under the aforementioned conditions. Peak purity report for HCTZ, AML and OLM are shown in Figure 2A-C. The purity plots reveal the absence of upslope, apex and downslope impurities. Moreover, the purity angels were much lower than the threshold values, revealing that the proposed method is specific for the assessment of the three drugs in their ternary therapy tablets.
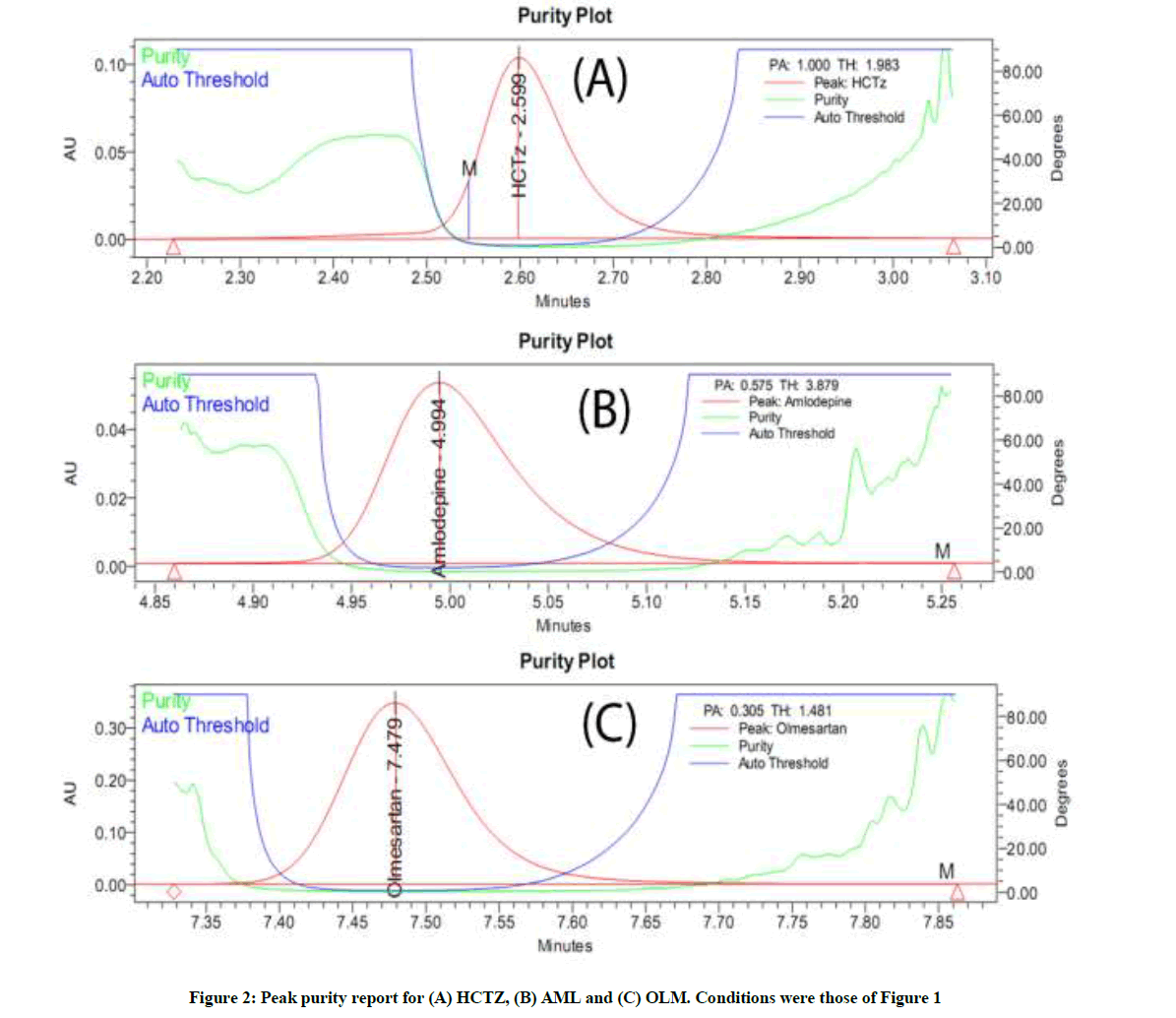
Figure 2: Peak purity report for (A) HCTZ, (B) AML and (C) OLM. Conditions were those of Figure 1
Linearity and range
Calibration curves representing the relation between drug concentration and peak area were linear over the investigated concentration ranges. The calibration parameters are shown in Table 1 that reveals the excellent linearity (R2 = 1.000) and reproducibility (RSD% < 2.0). In addition, Table 2 compares the developed RP-HPLC methods with some selected procedures revealing the superior performance of the proposed method regarding the calibration linearity, LOD, and affordability.
| Parameters | HCTZ | AML | OLM |
|---|---|---|---|
| Linear range (µg ml-1) | 0.12-17.50 | 0.05-7.00 | 0.20-28.00 |
| Regression Coefficient (R2) | 1 | 1 | 1 |
| Calibration slope | 50533.86 | 45039.47 | 90025.98 |
| Calibration intercept | 116.04 | -13.23 | 945.01 |
| SD of calibration curve | 181.235 | 29.938 | 568.745 |
| LOD (ng ml-1) | 12 | 2.2 | 21 |
| LOQ (ng ml-1) | 36 | 6.6 | 63 |
* Conditions were those of the recommended procedure
Table 1: Calibration parameters of HCTZ, AML and OLM*
| Method | HCTZ | AML | OLM | Ref | |||
|---|---|---|---|---|---|---|---|
| Range* | LOD* | Range* | LOD* | Range* | LOD* | ||
| UV | 2.00-12.00 | 0.022 | 1.00-6.00 | 0.028 | 3.00-18.00 | 0.024 | [3] |
| 5.00-35.00 | 10.00-80.00 | 10.00-60.00 | [4] | ||||
| 5.00-40.00 | 0.819 | 5.00-40.00 | 1.278 | 2.50-40.00 | 0.729 | [5] | |
| HPTLC | 0.50-3.00 | 0.162 | 0.50-3.00 | 0.188 | [6] | ||
| 0.50-2.50 | 0.058 | 0.20-1.00 | 0.009 | [7] | |||
| 0.025-0.15 | 0.006 | 0.08-0.48 | 0.018 | [8] | |||
| 1.00-3.50 | 0.325 | 1.00-3.50 | 0.303 | 0.80-1.80 | 0.33 | [9] | |
| HPLC | 2.50-100.00 | 0.17 | 5.00-100.00 | 0.59 | [10] | ||
| 0 | 0.10-50.00 | 0.1 | 0.10-50.00 | 0.1 | [11] | ||
| 0.002-2.50 | 0.002 | 0.008-10.00 | 0.008 | [12] | |||
| 15.00-250.0 | 1 | 25.00-500.00 | 1 | [13] | |||
| 5.48-41.07 | 2.17-16.31 | 9.06-67.92 | [14] | ||||
| 31.00-93.00 | 0.22 | 12.50-37.50 | 0.16 | 50.00-150.00 | 0.19 | [15] | |
| 44.00-82.00 | 17.00-32.00 | 70.00-130.00 | [16] | ||||
| 4.00-20.00 | 2.00-10.00 | 8.00-40.00 | 0.729 | [17] | |||
| UPLC | 1.25-3.75 | 0.50-1.50 | 2.00-6.00 | 0.19 | [18] | ||
| 1.50-30.00 | 0.213 | 2.00-27.00 | 0.469 | 10.00-60.00 | 0.308 | [19] | |
| 4.00-28.00 | 0.42 | 4.00-28.00 | 0.43 | [20] | |||
| 0.028 | 0.093 | [21] | |||||
| Current method | 0.12-17.50 | 0.012 | 0.05-7.00 | 0.002 | 0.20-28.00 | 0.021 |
* Expressed in μg ml-1
Table 2: Linear ranges and LOD of assay methods of HCTZ, AML and OLM
Accuracy and precision
The accuracy was tested by carrying out recovery studies at four spiking levels of 20, 60, 100 and 120% of the labeled amounts of HCTZ, AML and OLM in their combined ternary therapy (12.5/5/20 mg). Three replicate determinations were performed at each spiking level, Table 3. Percentage recoveries were 99.3-100.7% with RSD% of 0.20-0.07%, revealing the excellent accuracy of the proposed method. The precision of the developed method was further assessed for its reproducibility between runs and between two analysts. The between run and between analyst RSD% values were < 1.3 and 1.9%, confirming the high reproducibility of the proposed method.
| Theoretical %, µg mL-1 | Found µg mL-1 | Recovery (%) | SD | RSD (%) |
|---|---|---|---|---|
| HCTZ | ||||
| Sample 1 (20%), 2.5 | 2.483 | 99.3 | 293.87 | 0.23 |
| Sample 2 (60%), 7.5 | 7.48 | 99.74 | 1011.68 | 0.27 |
| Sample 3 (100%), 12.5 | 12.506 | 100.05 | 182.35 | 0.03 |
| Sample 4 (120%), 15.0 | 14.999 | 99.99 | 1053.57 | 0.14 |
| AML | ||||
| Sample 1 (20%), 1.0 | 1.005 | 100.46 | 110.49 | 0.24 |
| Sample 2 (60%), 3.0 | 2.994 | 99.81 | 148.56 | 0.11 |
| Sample 3 (100%), 5.0 | 4.998 | 99.97 | 164.71 | 0.07 |
| Sample 4 (120%), 6.0 | 6.041 | 100.69 | 41.04 | 0.02 |
| OLM | ||||
| Sample 1 (20%), 4.0 | 4 | 99.99 | 494.68 | 0.14 |
| Sample 2 (60%), 12.0 | 12.01 | 100.09 | 600.08 | 0.06 |
| Sample 3 (100%), 20.0 | 19.994 | 99.97 | 2272.17 | 0.13 |
| Sample 4 (120%), 24.0 | 24.003 | 100.01 | 463.64 | 0.02 |
Table 3: Results of accuracy and precision data
Stability of solution
Stability of the three drugs in solution was investigated and the analytical results show that the solutions are stable for 2 days at room temperature (15-25°C) and for at least 7 days at cool temperature (2-8°C) stored in amber colored flasks protected from light.
Robustness
The robustness of an analytical procedure is a measure of its insensitivity to small but deliberate changes in method conditions to indicate its reliability during normal usage. Robustness of the method was checked by analyzing the solutions used for accuracy and precision studies with small changes in column temperature (30 ± 2°C), Buffer pH (3.5 ± 0.05), flow rate (1.3 ± 0.1 ml min-1), wavelength (255 + 2 nm) and mobile phase composition (± 2%). Moreover, solution stability and filter interference were also assessed. Applying these deliberate changes resulted in quantitative drugs recovery for OLM, AML and HCTZ at the specified retention times with peak area differences of less than 2%. This reveals that the proposed method is robust as it successfully passed the test.
Dissolution profiles results
The developed RP-HPLC method was successfully used for determining the percentage drug released within 45 min for in-vitro dissolution of tablets containing the ternary therapy. The in-vitro dissolution studies revealed that ≥ 96% and ≥ 99% of labeled amounts of HCTZ, AML and OLM in their fixed combination tablets were released within 20 and 30 min, respectively, as shown in Figure 3. The dissolution pattern complies with the FDA Guidance standards indicating the suitability of the proposed RP-HPLC method for the dissolution release of the three drugs.
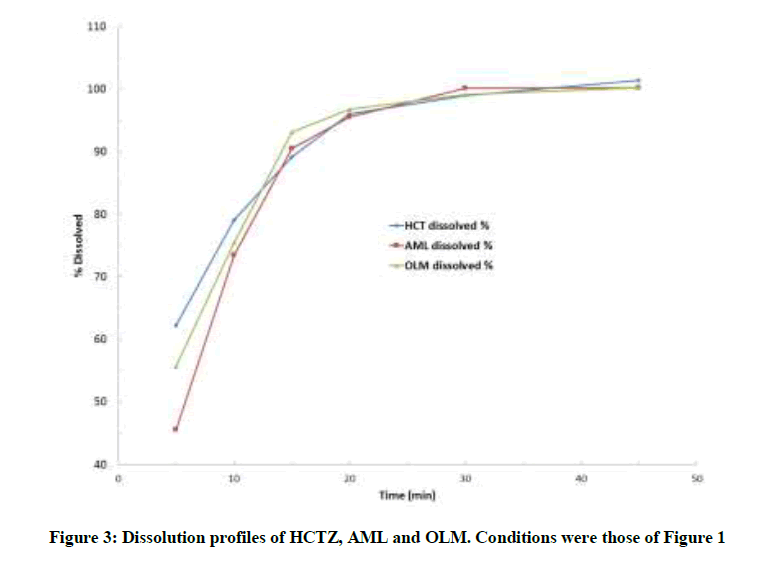
Figure 3: Dissolution profiles of HCTZ, AML and OLM. Conditions were those of Figure 1
Conclusion
In this study, a simple, highly sensitive and reliable RP-HPLC method was developed and validated for routine simultaneous assay and in vitro drug release studies of HCTZ, AML and OLM in their combined ternary therapy tablets. There were no interferences from blank, placebo and excipients at the retention times of HCTZ, AML or OLM. Peak purity results gave purity angles lower than the purity threshold indicating that the investigated drugs peaks were free from interference and revealed the specificity of the developed method. Moreover, the developed RP-HPLC method was validated, following the standard ICH guidelines, regarding specificity, suitability, linearity, accuracy, precision, and robustness. The proposed method showed superior sensitivity compared to existing methods [14-17] or the simultaneous assessment of HCTZ, AML or OLM in their ternary tablets.
References
- P. Bramlage, R. Ketelhut, E.M. Fronk, W.P. Wolf, R. Smolnik, C. Zemmrich, R.E. Schmieder, Clin. Drug. Investig., 2014, 34(6), 403-11.
- M. Volpe, A. de la Sierra, B. Ammentorp, P. Laeis, Adv. Ther., 2014, 31(5), 561-74.
- J. Saminathan, T. Vetrichelvan, INDIAN DRUGS., 2017, 54, 33-38.
- H.A. Merey, N.K. Ramadan, S.S. Diab, A.A. Moustafa, Spectrochim. Acta. A Mol. Biomol. Spectrosc., 2014, 125, 138-46.
- H.W. Darwish, Chem. Cent. J., 2013, 7, 22.
- K. Santhana Lakshmi, S. Lakshmi, J. Anal. Methods. Chem., 2012, 2012, 108281.
- B. Pankajbhai Marolia, K. Bharatbhai Bodiwala, S. Amritlal Shah, P. Bhagwanbhai Prajapati, B. Himmatbhai Satani, S. Alkeshbhai Desai, Pharmaceutical Methods., 2016, 7(1), 48-53.
- K. Ilango, P.S. Shiji Kumar, J. Anal. Methods. Chem., 2013, 2013, 363741.
- T.B. Solanki, P.A. Shah, K.G. Patel, Indian J. Pharm. Sci., 2014, 76(3), 179-187.
- Shaalan, R. A.; Belal, T. S., Gradient HPLC-DAD Determination of the Antihypertensive Mixture of Amlodipine Besylate, Valsartan, and Hydrochlorothiazide in Combined Pharmaceutical Tablets, J. Liq. Chromatogr. Rela. Technol., 2012, 35(2), 215-230.
- M.S. Kaynak, M. Celebier, S. Sahin, S. Altinoz, Revista De Chimie., 2013, 64(1), 27-30.
- S. Shah, A. Asnani, D. Kawade, S. Dangre, S. Arora, S. Yende, J. Young. Pharm., 2012, 4(2), 88-94.
- A.M. Brondi, J.S. Garcia, M.G. Trevisan, J. Anal. Methods. Chem., 2017, 2017, 4878316.
- V.D. Prasad, V. Rangareddy, P. Aparna, K.S. Babu, N.N. Prasad, Der Pharmacia Lettre., 2015, 7, 191-198.
- P.S. Jain, M.K. Patel, A.P. Gorle, A.J. Chaudhari, S.J. Surana, J. Chromatogr. Sci., 2012, 50(8), 680-687.
- A.A. Napoleon, G.A.R. Angajala, Der Pharmacia Lettre., 2015, 7, 182-196.
- G. Manoharan, M.A. Bratty, H.A. Alhazmi, Int. J. Pharmaceut. Sci. Rev. Res., 2016, 37, 205-209.
- K.K. Kumar, C.K. Rao, G. Madhusudan, K. Mukkanti, Am. J. Anal. Chem., 2012, 03(01), 50-58.
- H.A. Merey, N.K. Ramadan, S.S. Diab, A.A. Mostafa, Int. J. Adv. Sci. Eng. Technol., 2015, 3, 45-50.
- E. Dinc, Z.C. Ertekin, Talanta., 2016, 148, 144-152.
- J. Vojta, A. Jedlicka, P. Coufal, L. Janeckova, J. Pharm. Biomed. Anal., 2015, 109, 36-44.
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use 2005 ICH Harmonized Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology, Q2(R1), Complementary Guideline on Methodology, ICH, London, 1996.
- FDA, Dissolution Testing of Immediate Release Solid Oral Dosage Forms, U.S. Department of Health and Human Services Food and Drug Administration, Rockville, MD, 1997.
- Center for Drug Evaluation and Research, U.S. food and drug administration. Reviewer Guidance, Validation of chromatographic methods; FDA, Rockville, MD, 1994.



