Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 4
An Improved Process for the Preparation of Dabigatran Etexilate Mesylate and Synthesis of its Impurities
Sitaramaiah Devarasetty*, Ravi Janni, Rambabu Nunna and Raghu Ram Suraparaju
Vasudha Pharma Chem Limited, R & D Department, Parawada, Visakhapatnam, Andhra Pradesh, India
- *Corresponding Author:
- Sitaramaiah Devarasetty
Vasudha Pharma Chem Limited
R & D Department
Parawada, Visakhapatnam, Andhra Pradesh, India
Abstract
The present invention describes the improved process for the preparation of dabigatran etexilate mesylate and impurities synthesis. The Synthesis involves easily available commercial raw materials with Acylation from n-hexyl chloroformate makes to get desired Dabigatran Etexilate from the methane sulfonic acid it is finally as a dabigatran etexilate mesylate salt with an effective yield. Catalyst free and pressure handled reactions are not performed. Dabigatran is an effective oral prodrug of the thrombin (Factor IIa) inhibitor it has been approved by FDA and is widely used to reduce the risk of stroke in patients with non-valvular atrial fibrillation.
Graphical Abstract
Keywords
Dabigatran, Arterial fibrillation, Thrombosis and hexyl chloroformate
Introduction
Dabigatran Etexilate Mesylate (Pradaxa) [1] is an anticoagulant medication that can be taken by mouth widely used to reduce the risk stroke in patients with non-valular atrial fibrillation. An anticoagulation [2] factor can reduce the stroke by 60% whereas antiplatelet can reduce the risk stroke by 20%. The most widely used agents to reduce the strokes [3] are a dabigatran. The related mechanism in the anticoagulation factor related to the thrombin inhibitor.
The two distinguishing terms in the medical dictionary to identify the atrial fibrillation and non-valvular atrial fibrillation face the common problem for doctors to identify the strokes caused by atrial fibrillation and non-valvular atrial fibrillation. Atrial fibrillation is defined as irregular heart rhythm or abnormal heart rhythm whereas non-valular atrial fibrillation is defined as not caused by the valvular heart disease but caused by the atrial fibrillation with non-valular hence termed as non-valvular atrial fibrillation. The common symptoms of non-valvular atrial fibrillation are sleep apnea, high blood problems, lung problems, and hyperthyroidism [4]. The New medications are in using with non-valular atrial fibrillation are preferable to vitamin-K antagonists called as Novel Oral Anticoagulants (NOACS) are Dabigatran (Pradaxa), Rivaroxaban (Xarelto), Apixaban (Eliquis).
The intrinsic pathway mechanism of Pradaxa is a type of an anticoagulant, irregular function of heart leads to the effect on flow rate of blood and this can cause blood to pool in the upper chambers of your heart and this can be called as atria. The pooling can cause a clot to form in heart and this makes to travel directly to brain by blocking artery leads to stroke. Pradaxa is direct thrombin inhibitor factor (IIa) [5].
The present invention relates to the Pradaxa is chemically designated as β-alanine derivative and its IUPAC name is N-[[2- [[[4[[[(Hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5-yl]carbonyl]-N-2-pyridinyl-beta-alanineethylester methanesulfonate and its Empirical formula is C35H45N7O8S and it is shown in the below Figure 1.
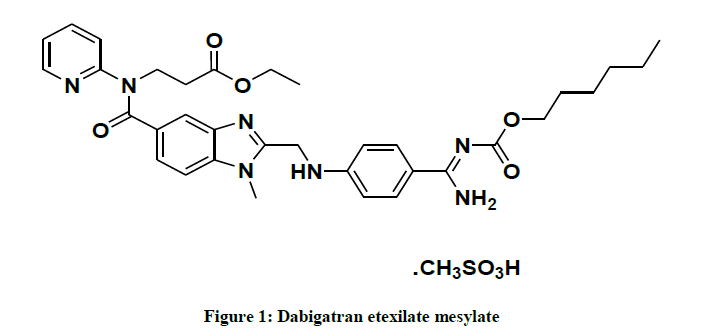
Figure 1: Dabigatran etexilate mesylate
According to the structural moiety [6] of dabigatran the presence of strongly basic amino group, it can be orally absorbed, the carboxylic ester and amido hexyl ester which leads a key role in converting the prodrug, play an important role in anti-coagulating effect. An oral anti-coagulant prevents fibrinogen lysis prevent fibrinin.
General Methods
Commercially available raw materials used without purification. Melting points and boiling points measured with an electronic melting apparatus and not corrected. 1H-NMR and 13C-NMR spectra recorded in CDCl3 as solvent on a 400 MHz Bruker spectrometer using Tetramethylsilane (TMS) as an internal standard and deuterium exchange also recorded. Chemical shifts calculated in terms of “ppm”. Infrared Absorption Spectrum (ABB-FT/IR spectrophotometer) is used for recording the spectrum with KBr disc technique on a 4000 cm-1 to 400 cm-1. The High-resolution Mass Spectrum (HRMS) was recorded using Shimadzu (Model LCMS-8030) triple quadruple mass spectrometer.
Experimental Procedures
Synthesis of ethyl-3-(2-((4-cyanophenylamino)methyl)-1-methyl-N-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamido)propanoate oxalate (III)
To a clean and dry RBF add dichloromethane (600.0 ml), 1,1-carbonylimidiazole (66.3 g, 0.409 mol) mixture to a 2-(4-cyanophenyl amino) acetic acid (61.6 g, 0.350 mol, II) slowly and stir for 30 min. Add ethyl-3-((3-amino-4-(methylamino)benzoyl)(pyridine-2-yl)amino)propanoate (100.0 g, 0.292 mol, I) in dichloromethane (300.0 ml) slowly to the above reaction mixture and stir for 8 h to complete the reaction. Monitor the completion of reaction by TLC. To the reaction mass, water (500.0 ml) was added and stir for 30 min. The organic and aqueous layers are separated. The organic layer is washed with water (500.0 ml) and concentrate under reduced pressure below 40°C to get the residue. To the residue ethyl acetate (600.0 ml) and acetic acid (100.0 ml) is added and reaction mixture is heated to 80-85°C and stir for 2 h. To the reaction mass, water (600.0 ml) is added and stir for 30 min. The organic and aqueous layers are separated. Wash the organic layer with 5% sodium bicarbonate solution (200.0 ml). Separate the organic layer and add oxalic acid dihydrate (35.0 g, 0.277 mol). Stir the reaction mixture for 1 h at 25 to 30°C. The isolated solid filtered, washed with ethyl acetate (100.0 ml) and dried to a constant weight at 60-65°C to yield ethyl 3-(2-((4- cyanophenylamino)methyl)-1-methyl-N-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamido)propanoate oxalate (III, yield 134.4 g, 80.5%).
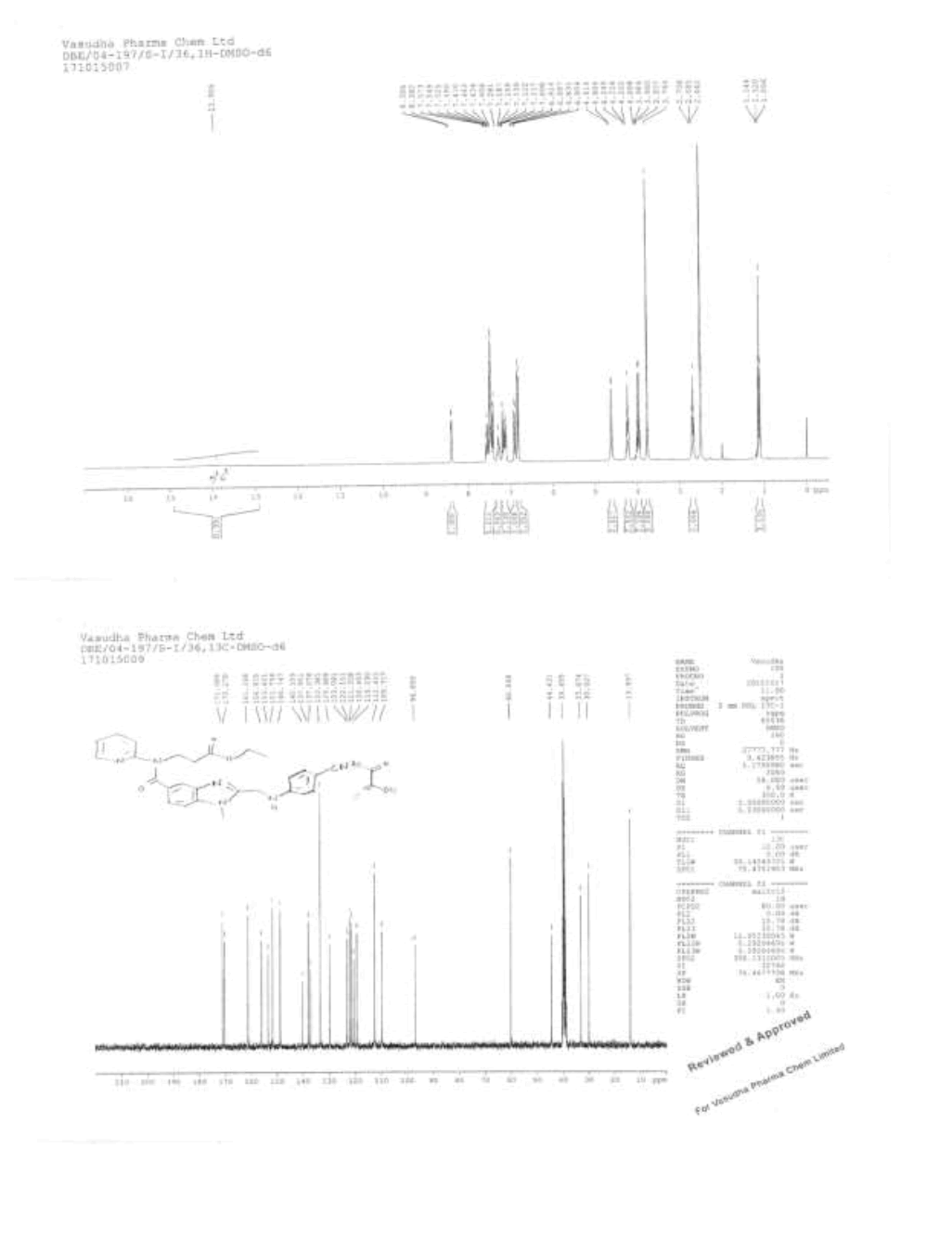
IR (cm-1): 3273 (-N-H stretching), 2939 (-C-H stretching in -Ar), 1728 (-C=O stretching in amide), 1753 (-C=O stretching in ester), 1568 (-C=C stretching in -Ar); 1H-NMR (CDCl3, δ ppm): 13.90 (s, 1H), 8.38 (d, 1H), 7.55 (t, 1H), 7.52-7.40 (m, 4H), 7.281 (s, 1H), 7.18-7.09 (m, 2H), 6.89 (d, 1H), 6.81 (d, 2H), 4.60 (s, 2H), 4.22 (t, 2H), 4.08-3.93 (m, 2H), 3.76 (s, 3H), 2.68 (t, 2H), 1.11 (t, 3H); 13C-NMR (CDCl3, δ ppm): 171.08, 170.27, 161.26, 156.0, 153.42, 151.75, 148.74, 140.15, 137.95, 137.07, 133.38, 129.68, 123.09, 122.15, 121.32, 120.45, 119.29, 112.45, 109.71, 96.85, 60.06, 44.43, 39.65, 33.07, 30.0, 13.99; Mass: 483.4 (M+)-Free Base.
Synthesis of ethyl-3-(2-((4-amidinophenylamino)methyl)-1-methyl-N-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamido)propanoate hydrochloride (IV)
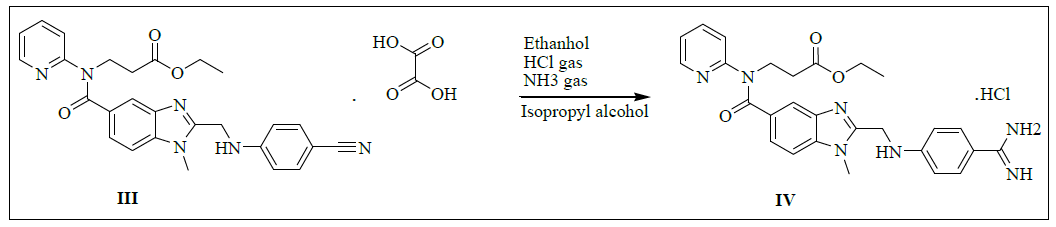
To a mixture of 3-(2-((4-cyanophenylamino)methyl)-1-methyl-N-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamido)propanoate oxalate (100.0 g, 0.174 mol, III) and ethanol (1000.0 ml) added slowly passed to the dry HCl gas get 35-40% and stirred for 8 h to complete the reaction. Monitor the completion of reaction by TLC and concentrated under reduced pressure below 40°C to get the residue. To the residue Ethanol (1000.0 ml) added and adjusted pH: 9-10 by using dry ammonia gas and stirred for 10 h to complete the reaction. Distill off Ethanol completely under reduced pressure below 50°C to get the residue. To the residue cyclohexane (600.0 ml) is added and stirred for 30 min. isolate the solid filtrate and washed with cyclohexane 100.0 ml). Take wet material and add Isopropyl alcohol (600.0 ml) and water (45.0 ml) and heat up to 80- 85°C. Isolate the Filtrate and dry to a constant weight at 65-70°C to yield ethyl 3-(2-((4-amidinophenylamino)methyl)-1-methyl-N-(pyridin-2- yl)-1H-benzo[d]imidazole-5-carboxamido)propanoate hydrochloride (IV, yield 80.4 g, 86.4%).
IR (cm-1): 3271 (-N-H stretching), 2978 (-C-H stretching in -Ar) 1634-1645 (-C=N stretching), 1730 (-C=O stretching), 1587 (-C=C stretching in -Ar); 1H-NMR (CDCl3, δ ppm): 8.90 (s, 2H), 8.69 (s, 2H), 8.38 (d, 1H), 7.66 (d, 2H), 7.55 (t, 1H), 7.47-7.39 (m, 3H), 7.16 (t, 2H), 6.87 (t, 3H), 4.64 (d, 2H), 4.22 (t, 2H), 4.0 (t, 2H), 3.77 (s, 3H), 2.67 (t, 2H), 1.11 (t, 3H); 13C-NMR (CDCl3, δ ppm): 171.05, 170.33, 164.44, 155.99, 153.43-153.07, 148.70, 140.79, 137.92-153.07, 148.0, 140.79, 137.92-137.24, 129.65-129.36, 122.81-122.10, 121.29, 119.51, 113.19, 111.70, 109.53, 60.03, 44.37, 33.05, 30.0, 25.49, 13.97; Mass: 536.03 (M+).
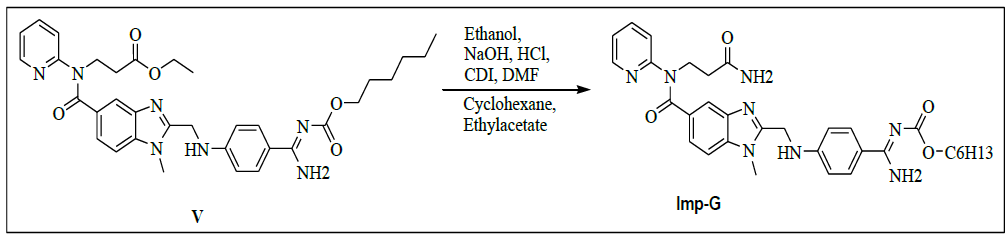
Synthesis of ethyl-3-[[[2-[[[[[(hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5- yl]carbonyl](pyridin-2-yl)amino]propanoate (V)
A mixture of ethyl-3-(2-((4-amidinophenylamino)methyl)-1-methyl-N-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamido)propanoate hydrochloride (100.0 g, 0.186 mol, IV) and acetone (1000.0 ml), potassium carbonate (154 g, 1.114 mol) and stirred for 30 min. Hexyl chloroformate (42.2 g, 0.256 mol) in acetone (200.0 ml) was added slowly to the reaction mass and stirred for 2 h to complete the reaction. Monitor the completion of reaction by HPLC. To the reaction mass water was added and stirred for 30 min. The organic and aqueous layers were separated. Distill off acetone completely under reducing pressure below 50°C to get a residue. To the residue, Isopropyl alcohol (1500.0 ml) was added and stirred for 2 h. Isolate the solid filtrate and wash with isopropyl alcohol (200.0 ml) and dried to the constant weight at 55-60°C to yield ethyl-3-[[[2-[[[[[[(hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5-yl]carbonyl](pyridin-2- yl)amino]propanoate (V, 87.8 g, 75%).
IR (cm-1): 3408 (-N-H stretching), 3381 (-C-H stretching in -Ar), 1730 (-C=O stretching), 1612-1655 (-C=N stretching), 1570 (-C=C- stretching in -Ar), 1286 (-C-O stretching in ester); 1H-NMR (CDCl3, δ ppm): 8.91 (s, 2H), 8.38 (d, 1H), 7.79 (d, 2H), 7.54 (d, 2H), 7.47 (s, 1H), 7.39 (d, 1H), 7.17-7.09 (m, 2H), 6.96 (t, 1H), 6.88 (d, 1H), 6.76 (d, 2H), 4.59 (d, 2H), 4.22 (t, 2H), 4.0-3.9 (m, 4H), 3.76 (s, 3H), 2.68 (t, 2H), 1.60-1.53 (m, 2H), 1.286 (s, 6H), 1.11 (t, 3H), 0.86 (t, 3H); 13C-NMR (CDCl3, δ ppm): 171.02, 170.32, 166.40, 164.24, 156.01, 153.70, 151.56, 148.67, 140.83, 137.85, 137.24, 129.29-129.16, 122.79, 122.09, 121.22, 121.05, 119.51, 111.36, 109.44, 64.08, 44.34, 39.91, 33.02, 31.01, 29.89, 28.53, 25.23, 22.05, 13.94, 13.89; Mass: 628.5 (M+).
Synthesis of N-[[2-[[[4-[[[(Hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5-yl]carbonyl]-N-2- pyridinyl-β-alanineethylester methanesulfonate (VI)
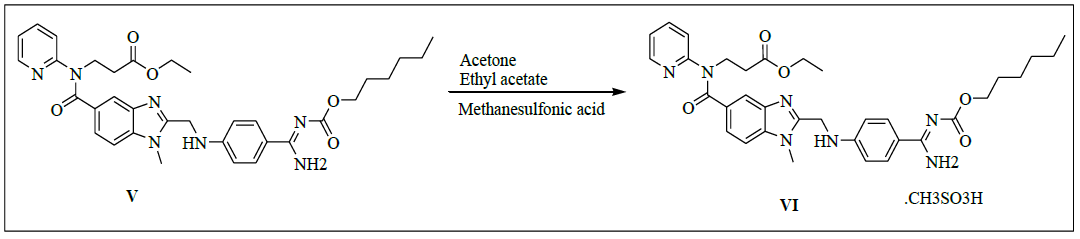
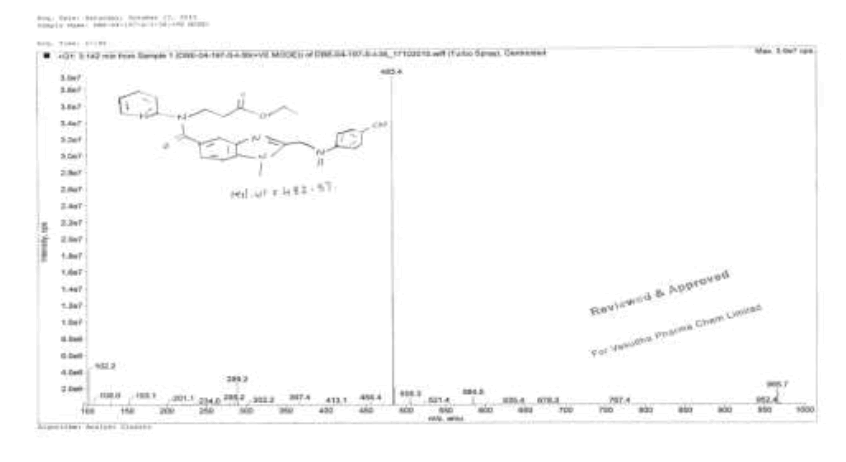
To a mixture of ethyl-3-[[[2-[[[[[[(hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5- yl]carbonyl](pyridin-2-yl)amino]propanoate (100.0 g, 0.159 mol, V) and acetone (1000.0 ml) was added and stirred for 15 min. Filter the reaction mixture through hi-flow bed. Methanesulfonic acid (15.3 g, 0.159 mol) in ethyl acetate (400.0 ml) was added slowly to the reaction mass and stirred for 2 h to complete the reaction. Isolate the solid filtrate and washed with Acetone (200.0 mL), and dried to the constant weight below 50°C under reduced pressure to yield N-[[2-[[[4-[[[(Hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1H-benzimidazol- 5-yl]carbonyl]-N-2-pyridinyl-β-alanineethylester methanesulfonate (VI, Yield 109.5, 95%).
IR (cm-1): 3263 (N-H stretching), 2957 (-C-H stretching in -Ar), 1610-1645 (-C=N stretching), 1734 (-C=O stretching), 1587 (-C=C-stretching in -Ar), 1288 (-C-O stretching in -COOR); 1H-NMR (CDCl3, δ ppm): 11.25 (s, 2H), 10.01 (s, 1H), 8.38 (d, 1H), 7.63 (d, 3H), 7.55 (t, 1H), 7.44 (t, 2H), 7.17-7.10 (m, 2H), 6.88 (t, 3H), 4.69 (t, 2H), 4.27-4.19 (m, 4H), 4.27-4.19 (m, 4H), 4.06-3.93 (m, 2H), 3.779 (s, 3H), 2.67 (t, 2H), 2.29 (s, 3H), 1.72-1.63 (m, 2H), 1.39-1.29 (m, 6H), 1.11 (t, 3H), 0.87 (t, 3H); 13C-NMR (CDCl3, δ ppm): 171.03, 170.24, 163.43, 155.96, 154.09, 153.79, 153.27, 148.70, 140.34, 137.93, 137.11, 131.40, 129.56, 122.95, 122.09, 121.31, 119.30, 112.44, 111.70, 109.66, 66.90, 66.01, 44.36, 39.74, 33.09, 30.83, 30.0, 27.95, 24.82, 22.0, 13.97, 13.88; Mass: 628.5 (M+).
Results and Discussion
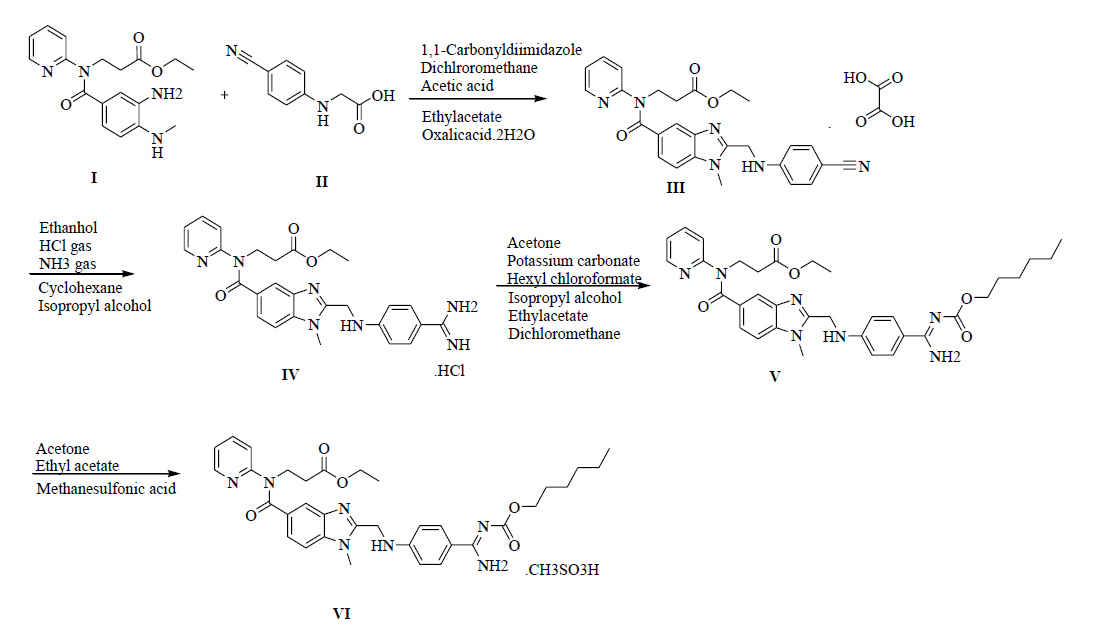
The improved synthesis involves with the commercially available starting material ethyl-3-(3-amino-4-(methylamino)benzoyl(piperidin-2- yl)amino)propante (I), condensation with 2-cyanophenyl amino acetic acid (II), and on oxalic acid resulting in the formation of ethyl 3-(2-((4- cyanophenylamino)methyl)-1-methyl-N-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carbox amido)propanoate oxalate salt (III) as an off-white with an yield of 80%. In the presence of HCl gas by treating with oxalate salt yields to form a Ethyl 3-(2-((4-amidinophenylamino)methyl)-1-methyl- 1-N-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamido)propanate hydrochloride salt (IV), as a white solid with an yield of 85%. The acylation reaction with hexyl chloroformate with the obtained hydrochloride salt makes to get the desired compound as ethyl-3- [[[(hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5-yl]carbonyl](pyridin-2-yl)amino]propanoate (V) as a white powder with an yield of 80%. Treatment with methane sulfonic acid in acetone to obtain the final compound Dabigatran Etexilate Mesylate salt [7-10] (V1) with a yield of 98%.
The dabigatran etexilate mesylate is first reported in 98/37035. In this they disclosed 1-methyl-2-[N-[4-N-hexyloxycarbonylamidino) phenyl]amniomethyl]benzoimidazol-5-yl-carboxylic acid-N-(2-pyridyl)-N-(2-ethoxycarbonylethyl)amide (Dabigatran etexilate) [11]. The conversion of salt form is not disclosed. The process of mesylate salt and its polymorphic forms was disclosed in US2005/234104. In this methane sulfonic acid and acetone were used to obtain finally as Dabigatran Etexilate Mesylate with a purity of 97-98% by HPLC is not a satisfactory result.
In some Embodiments WO2009/153214 the reduction of nitro to amine through catalytic hydrogenation in the presence of tertiary amine under hydrogen pressure involves pressure reactions and recovery of the palladium on charcoal catalyst, which are difficult at large scale production. In WO2012/004397 the use of inorganic base under hydrogen pressure leads to problems which require a skill and art to overcome this in the sense of purity and good yield. Pressure controlling reactions are not monitored in large scale batches. The catalytic performance reactions are not suitable for commercial scale due to catalytic poisoning which leads to incomplete reaction.
All previous reports have some drawbacks like yield or purity or commercial scale reproducibility, whereas present invention has overcome all the issues and able produce from commercially available raw materials.
The synthesis in this present improved [12] process with high yields in low costs with easy availability of commercial chemicals without involvement of catalyst and pressure conditions. There was tremendous progress in the synthesizing with elimination of potential impurities. The effective overall yield comparative to the above invention has good results with qualitative and quantitative purity in large scale industrial process which is used for large scale commercializing.
As it is very important in the process of characterization of pure form of drug according to ICH [13,14] guidelines to review and identify the impurity in the drug. The different pharmacopoeias are slowly incorporating limits that to allowable levels in the API’s. As apart from the past inventions and now in the present inventions the impurities [15] and its detection play a key role in the purity of drugs. It is extremely challenging to identify the impurities [16-18] that formed in very minor quantities and had to be traced in drug substances. In this present invention we synthesize the preparation of Pradaxa (VI) and it is shown in Scheme 1 and their impurities synthesis in Scheme 2 and their structures with IUPAC names are Table 1.
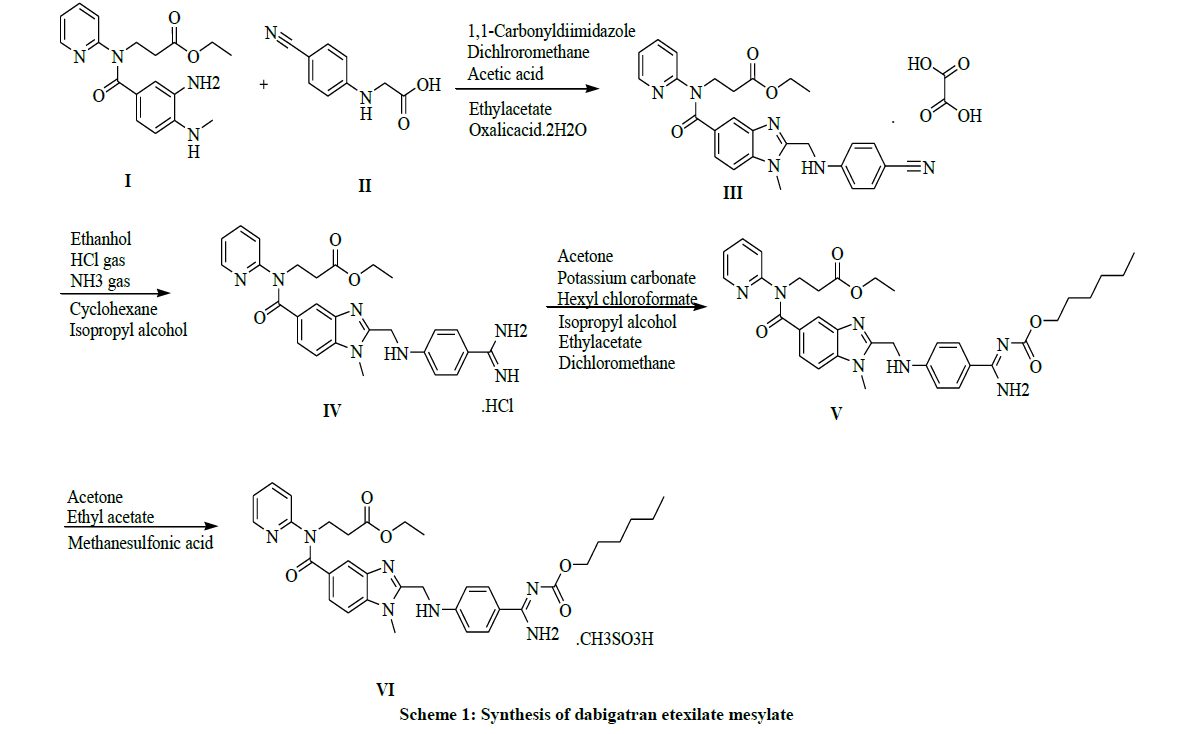
Scheme 1: Synthesis of dabigatran etexilate mesylate
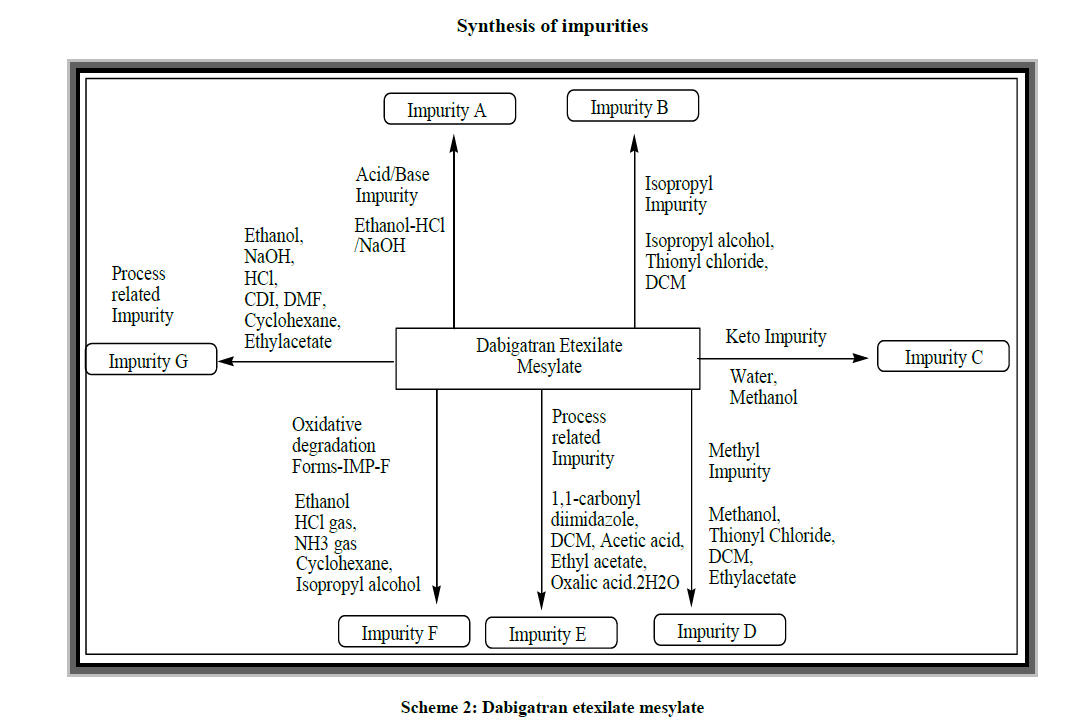
Scheme 2: Dabigatran etexilate mesylate
| S. No. | Name of the compound | Structure | CAS Number |
|---|---|---|---|
| 1 | N-[[2-[[[4-[[ (Hexyloxy) carbonyl]amino]iminomethyl]phenyl] amino] methyl]-1-methyl-1H-benzimidazol-5-yl] carbonyl]-N-2-pyridinyl-b-alanine ethyl ester methane sulfonate. | 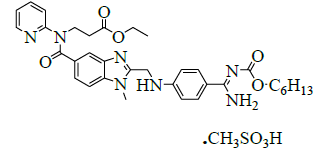 |
872728-81-9 |
| 2 | Ethyl-3-[[[2-[[[4-[[[ (hexyloxy)carbonyl]amino] iminomethyl]phenyl]amino]methyl]-1-methyl-1H- benzimidazol-5-yl] carbonyl] (pyridin-2-yl) amino] propanoate [N-1 stage]. | 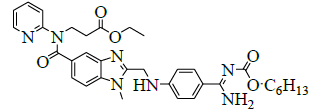 |
211915-06-9 |
| 3 | 3-{[ (2-{[ (4-{[ (hexyloxy) carbonyl]carbamimidoyl}phenyl)amino] methyl}-1-methyl-1H-benzimidazo-1-5-yl) carbonyl] (pyridin-2-yl) amino} propanoicacid [IMP-A]. | 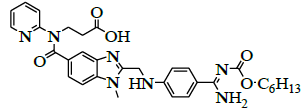 |
212321-78-3 |
| 4 | Propyl-3-{[ (2-{[ (4-{[ ({hexyloxy} carbonyl)amino]carbonyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl] (pyridin-2-yl)amino}propanoate [IMP-B]. | 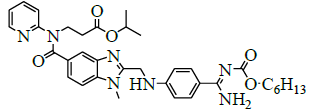 |
1610758-19-4 |
| 5 | Ethyl-3-{[ (2-{[ (4-{[ ({hexyloxy}carbonyl)amino]carbonyl} phenyl) amino] methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl] (pyridin-2-yl)amino}propanoate [IMP-C]. | 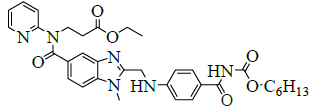 |
1408238-40-3 |
| 6 | Methyl-3-{[ (2-{[ (4-{[ (hexyloxy)carbonyl] carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl] (pyridin-2-yl)amino}propanoate [IMP-D]. |
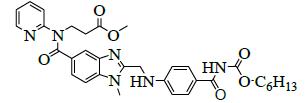 |
211915-00-3 |
| 7 | Ethyl-3- (2- ( (4-cyanophenylamino) methyl)-1-methyl-N- (pyridin-2-yl)-1H-benzo[d] imidazole-5-carboxamido)propanoate oxalate [IMP-E]. | 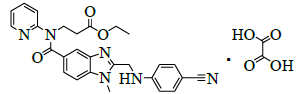 |
1223061-77-5 |
| 8 | Ethyl-3- (2- ( (4-amidinophenylamino)methyl)-1-methyl-N- (pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamido)propanoate hydrochloride [IMP-F]. | 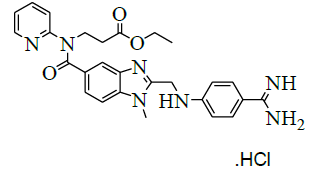 |
211914-50-0 |
| 9 | N-[[4-[[[5-[[ (3-amino-3-oxopropyl)-2-pyridinylamino]carbonyl]-1-methyl-1H-benzimidazol-2-yl]methyl]amino]phenyl]iminomethyl]-carbamic acid hexyl ester [IMP-G]. | 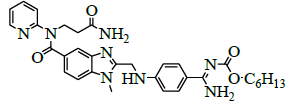 |
1580491-16-2 |
Table 1: The impurities structures and there IUPAC names
As of our keen interest of investigation to isolate the pure form of dabigatran we identify the impurities listed in tabulated as follows:
Impurities of dabigatran etexilate mesylate
Impurity A: The impurity formed from the process related and degradants impurity identified during the synthesis. Metabolite impurity, acidic hydrolysis or basic hydrolysis leads to the formation of Impurity A.
Impurity B: The impurity is formed from the process related and this can be identified by the traces amount of an isopropyl alcohol during synthesis leads to the evaluation of Impurity B.
Impurity C: Keto Impurity formed from the process related and degradants impurity due to temperature and humidity conditions forms an Impurity C.
Impurity D: The impurity is formed from the process related and this can be identified by the traces amount of a methanol during synthesis and this form the Acid/Base in presence of water leads to the evaluation of Impurity D.
Impurity E: This is formed from the process related impurity.
Impurity F: Oxidative degradation of dabigatran leads to the formation Impurity F. This is formed from the process related and metabolic impurity.
Impurity G: The impurity formed from the process related and degradants impurity.
Conclusion
We have developed a convenient efficient process for the preparation of dabigatran etexilate mesylate and synthesis of its impurities. We can easily manufacture dabigatran etexilate mesylate with this route in large scale by using commercially available raw materials in with high yields.
Acknowledgement
The authors are thankful to Vasudha Pharma Chem Limited for their support and are also thankful to Mr. chiranjeevi varma for drafting manuscript.
References
- A.J. Awad, B.P. Walcott, C.J. stapleton, V. Yanamandala, B.V. Nahed, J.V. Coumans, Neurosurg Focus., 2013, 34, 1.
- T.J. Moore, M.R. Cohen, D.R. Mattison, BMJ., 2014, 349, g4517.
- N.H. Hauel, H. Nar, H. Pripke, U. Ries, J.-M. Stassen, W. Wienen, J. Med. Chem., 2002, 45, 1757.
- S. Sinare, S. Borhade, S. Jadhav, V. Acharya, P.V. Srinivasan, Int. J. Pharm. Res. Dev., 2014, 6, 119.
- J. Van Ryn, J. Stangier, S. Haertter, K.H. Liesenfeld, W. Wienen, M. Feuring, A. Clemens, Thromb. Haemost., 2010, 103, 1116.
- N.H. Schemmerhofen, W. Henning Priepke, B. Uwe Ries, Patent-US6087380.
- Y.Y. Zheng, C. Shen, M. Zhu, Y. Zhou, J. Li, Org. Process Res. Dev., 2014, 18(6), 744-750.
- D. Mohan Rao, A. Jithender, Patent-WO2015132794.
- P. Laxmikant Narhari, M. Harish Kasinath, Patent-WO2014167577.
- B. Ravindra Babu, N. Subha Velayudhan, Patent-WO2014192030.
- N. Hauel, U. Ries, H. Priepke, W. Wienen, J. Stassen, Patent-1998037075 A1.
- S. Eswaraiah, A. Ramprasad, C. Nagaraju, S. Raghuram, S. Satyanarayana, Patent-WO2012077136.
- FDA Drug Safety Communication: (Dabigatran Etexilate Mesylate) should not be used in patients with Mechanical Prosthetic Heart Valves, U.S. Food and Drug Administration (FDA), 2014.
- ICH Q3A Impurities in New Drug Substances, R2; International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human use (ICH): Geneva, Switzerland, October, 2006.
- S.D. Khasim Sharif, S. Sri Ramudu, N. Rambabu, N. Ramchandran, Asian J. Chem., 2017, 29(6), 1253-1257.
- U. Rajasekhar Reddy, B. Sreenivasulu, D. Satheesh, T. Dhanraj, S. Sundaram, G. Srikanth, S. Paul Douglas, Synth. Commun., 2017, 47(13), 1225-1230.
- http://dx.doi.org/10.1016/j.arabjc.2015.09.006.
- S. Norbert Hauel, H.W. Priepke, B. Uwe Ries, Patent-US6087380.
Supplementary Data
Experimental procedure for the synthesis of impurities
Synthesis of 3-{[(2-{[(4-{[(hexyloxy)carbonyl]carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazo-1-5-yl) carbonyl](pyridin-2-yl)amino}propanoic acid (Impurity A):
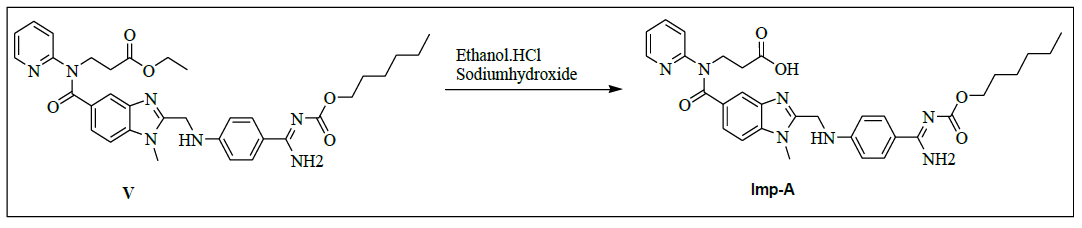
To a mixture of ethyl-3-[[[2-[[[[[[(hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5- yl]carbonyl](pyridin-2-yl)amino]propanoate (50.0 g, 0.079 mol, V) and ethanol (500.0 mL), 10% sodium hydroxide solution (50.0 ml) added then reaction mixture is stirred for 2 h. Monitor the reaction by TLC, after completion. Distill off ethanol completely under reduced pressure below 50°C. To the reaction mixture, water added and adjusted pH 4-5 with 10% Con. Hydrochloric acid (21.2 ml) and stirred the reaction mass for 1 h at 25-30°C. Isolated the solid filtrate and washed with water (200.0 ml) and dried to the constant weight below 50°C under reduced pressure to yield 3-{[(2-{[(4-{[(hexyloxy)carbonyl]carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazo-1-5-yl)carbonyl](pyridin- 2-yl)amino}- propionic acid (IMP-A, Yield 43.0 g, 90%).
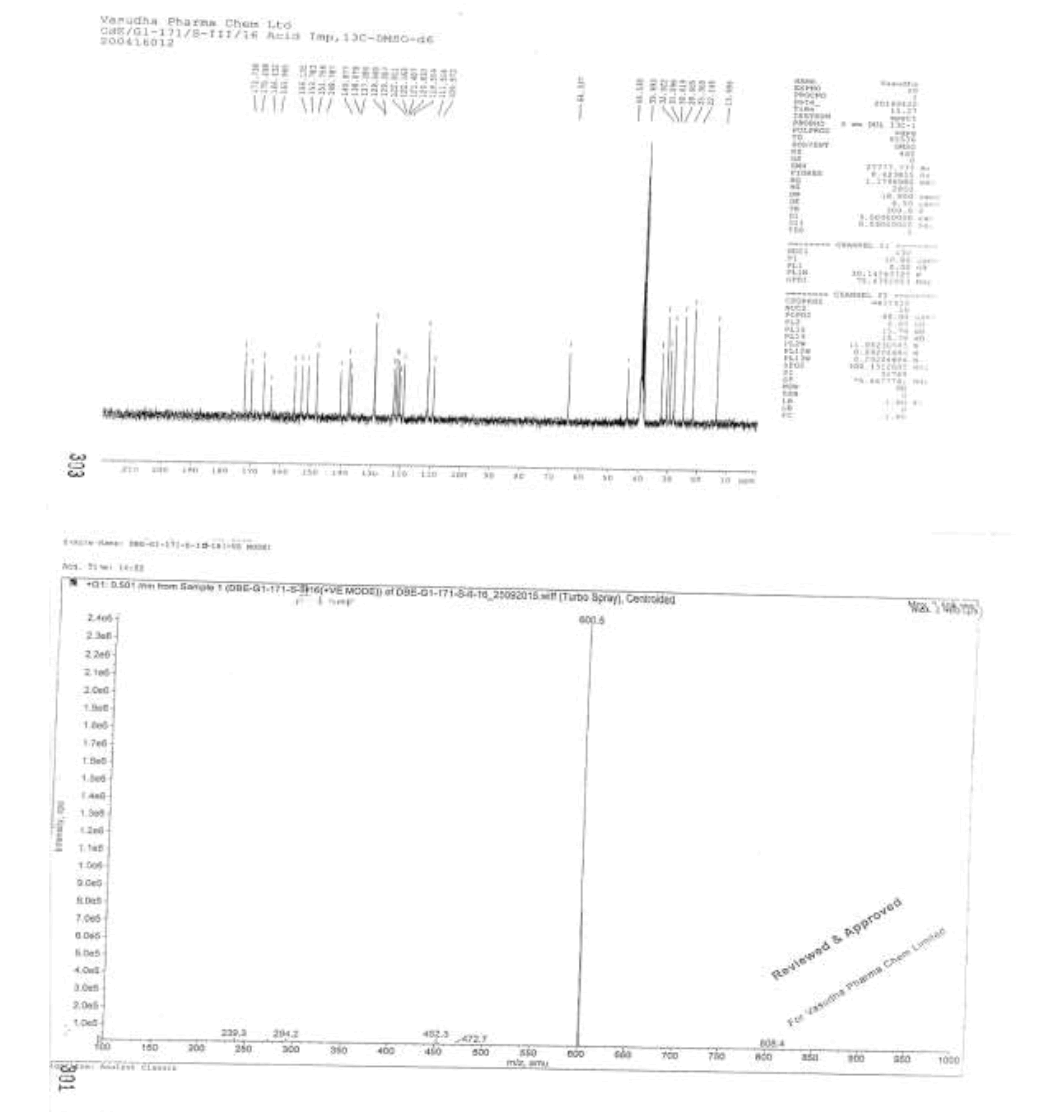
IR (cm-1): 3315 (-O-H stretching), 2955 (-C-H stretching in -Ar), 1732 (-C=O stretching), 1609-1645 (-C=N stretching), 1589 (-C=C-stretching in -Ar), 1283 (-C-O Stretching in -COOR); 1H-NMR (CDCl3, δ ppm): 12.28 (s, 1H), 8.91 (s, 2H), 8.37 (d, 1H), 7.78 (d, 2H), 7.55 (t, 1H), 7.48 (s, 1H), 7.39 (d, 1H), 7.17-7.10 (m, 2H), 6.95 (t, 2H), 6.76 (d, 2H), 4.59 (d, 2H), 4.17 (t, 2H), 3.95 (t, 2H), 3.76 (s, 3H), 2.61 (t, 2H), 1.57 (d, 2H), 1.28 (s, 6H) 0.86 (d, 3H); 13C-NMR (CDCl3, δ ppm): 172.73, 170.49, 166.43, 163.96, 156.13, 153.78, 151.75, 148.78. 140.87, 138.07, 137.28, 129.56, 129.35, 122.91, 122.16, 121.40, 120.83, 119.55, 111.54, 109.57, 64.33, 44.53, 39.99, 32.92, 31.09, 30.0, 28.60, 25.30, 22.14, 13.0; Mass: 600.5 (M+).
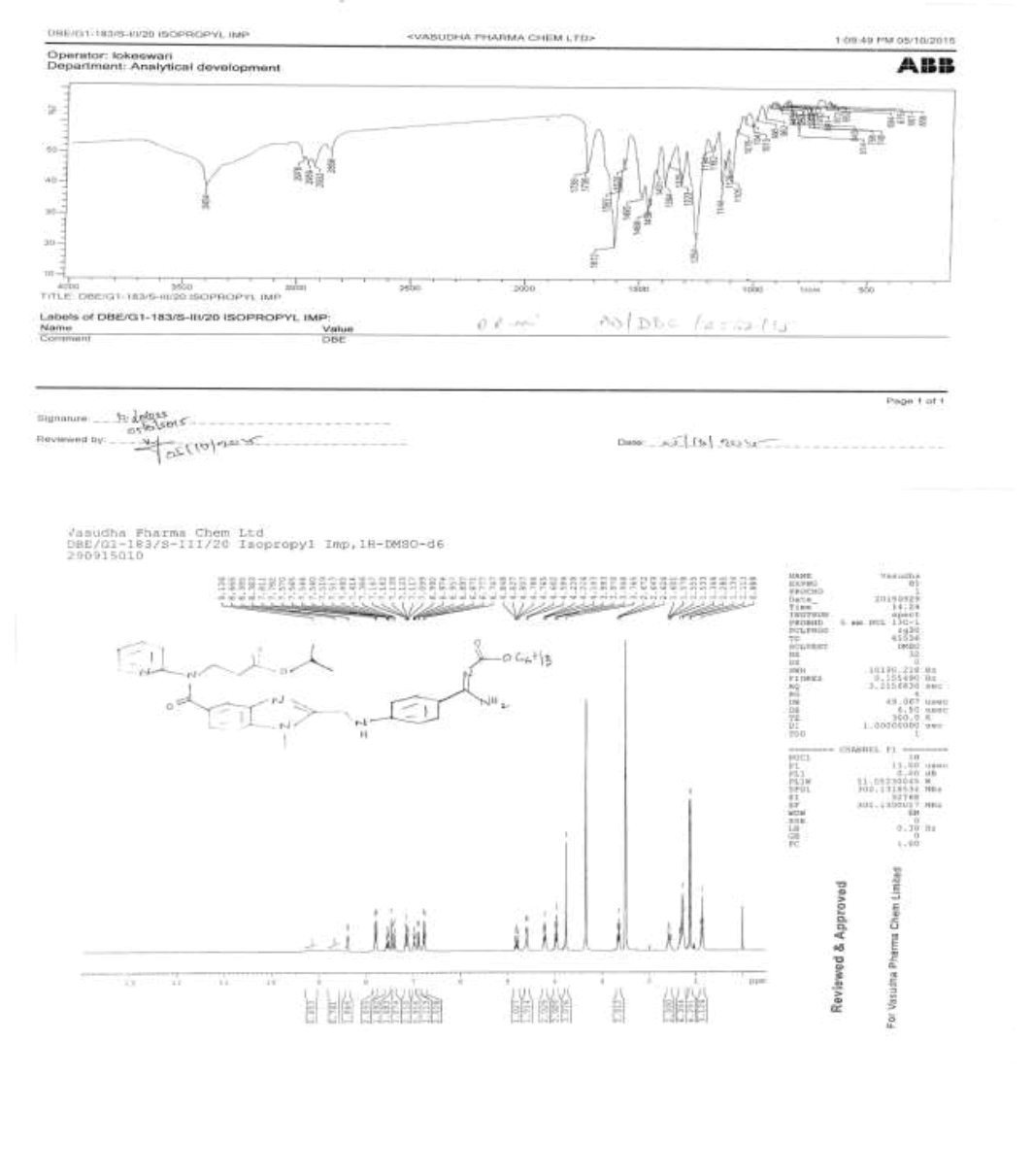
Synthesis of propyl-3-{[(2-{[(4-{[({hexyloxy} carbonyl) amino] carbonyl} phenyl) amino] methyl} - 1- methyl- 1H-benzimidazol-5-yl) carbonyl] (pyridin-2-yl) amino} propanoate (Impurity B):
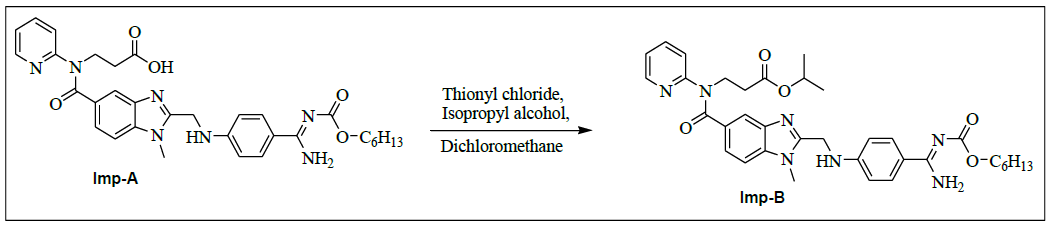
To a mixture of ethyl-3-[[[2-[[[[[[(hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5- yl]carbonyl](pyridin-2-yl)amino]propanoate (50.0 g, 0.079 mol, V) and ethanol (500.0 ml), 10% sodium hydroxide solution (50.0 ml) added, then reaction mixture stirred for 2 h. Monitor the reaction by TLC, once after completion distill off ethanol completely under reduced pressure below 50°C. To the reaction mixture water added and adjusted pH to 4-5 with 10% Con. Hydrochloric acid (21.2 ml) and stirs the reaction mass for 1 h at 25-30°C. Filtered reaction mass, washed with water (200.0 ml), and dried to the constant weight below 50°C under reduced pressure to yield 3-{[(2-{[(4-{[(hexyloxy)carbonyl] carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2- yl)amino}propanoicacid (IMP-A, Yield 43.0 g, 90%).
To the mixture of 3-{[(2-{[(4-{[(hexyloxy)carbonyl]carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazo-1-5- yl)carbonyl](pyridin-2-yl)amino}propanoic acid (50.0 g, 0.083 mol, IMP-A) and isopropyl alcohol (250.0 ml), the reaction mixture stirred for 30 min. Thionyl chloride (19.8 g, 0.166 mol) added to the reaction mixture below 20°C. Stir the reaction mixture for 2 h at 25-30°C. Monitor reaction by TLC. Once after completion, distill off the reaction mixture with isopropyl alcohol and thionyl chloride under reduced pressure below 50°C. Added water (300.0 ml) to the reaction mixture and adjusted pH to 8-9 with 10% sodium carbonate solution (45.0 ml) and extracted with Dichloromethane (300.0 ml). Distill off dichloromethane completely the organic layer under reduced pressure below 50°C to get the residue. Ethyl acetate (350.0 ml) added to the residue and stirred for 30 min. The isolated solid filtered and washed with ethyl acetate (100.0 ml) and dried to the constant weight below 50°C under reduced pressure to yield propyl-3-{[(2-{[(4- {[({hexyloxy}carbonyl)amino]carbonylphenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2-yl)amino}propanoate (IMP-B, Yield 49.7 g, 93%).
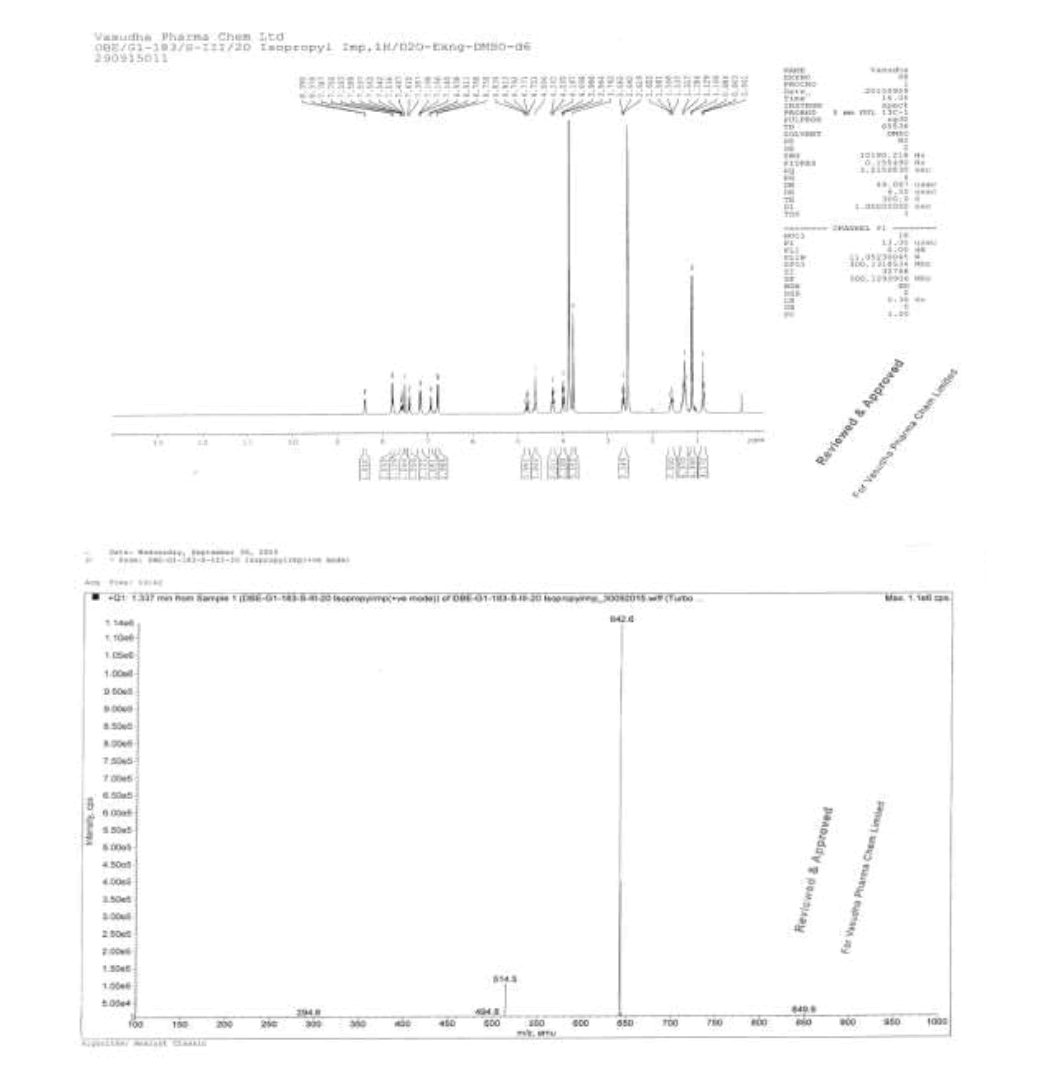
IR (cm-1): 3404 (-NH stretching), 2978 (-C-H stretching in aromatic), 1738 (-C=O stretching in ester), -1736 (-C=O Stretching in amide), 1587- 1612 (-C=N stretching), 1570 (-C=C- stretching in amide), 1587-1612 (-C=N stretching), 1570 (-C=C-stretching in aromatic), 1323 (-C-O stretching in isopropyl ester); 1H-NMR (CDCl3, δ ppm): 8.89 (s, 2H), 8.38 (d, 1H), 7.79 (d, 2H), 7.57-7.51 (m, 1H), 7.46 (s, 1H), 7.39 (d, 1H), 7.16-7.09 (m, 2H), 6.97 (t, 1H), 6.88 (d, 1H), 6.75 (d, 2H), 4.84-4.76 (m, 1H), 4.59 (d, 2H), 4.21 (t, 2H), 3.96 (t, 2H), 3.76 (s, 3H), 2.64 (t, 2H), 1.60-1.53 (m, 2H), 1.31 (s, 6H), 1.12 (d, 6H), 0.86 (t, 3H); 13C-NMR (CDCl3, δ ppm): 170.53, 170.29, 166.39, 164.22, 155.99, 153.70, 151.56, 148.68, 140.81, 137.88, 137.23, 129.30, 129.15, 122.78, 122.13, 121.25, 121.0, 119.50, 111.35, 109.44, 67.34, 64.07, 44.28, 39.90, 33.27, 31.0, 29.90, 28.53, 25.22, 22.05, 21.49, 13.89; Mass: 642.6 (M+).
Synthesis of ethyl-3-{[(2-{[(4-{[({hexyloxy}carbonyl)amino]carbonyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl) carbonyl] (pyridin-2-yl) amino} propanoate (Impurity C):
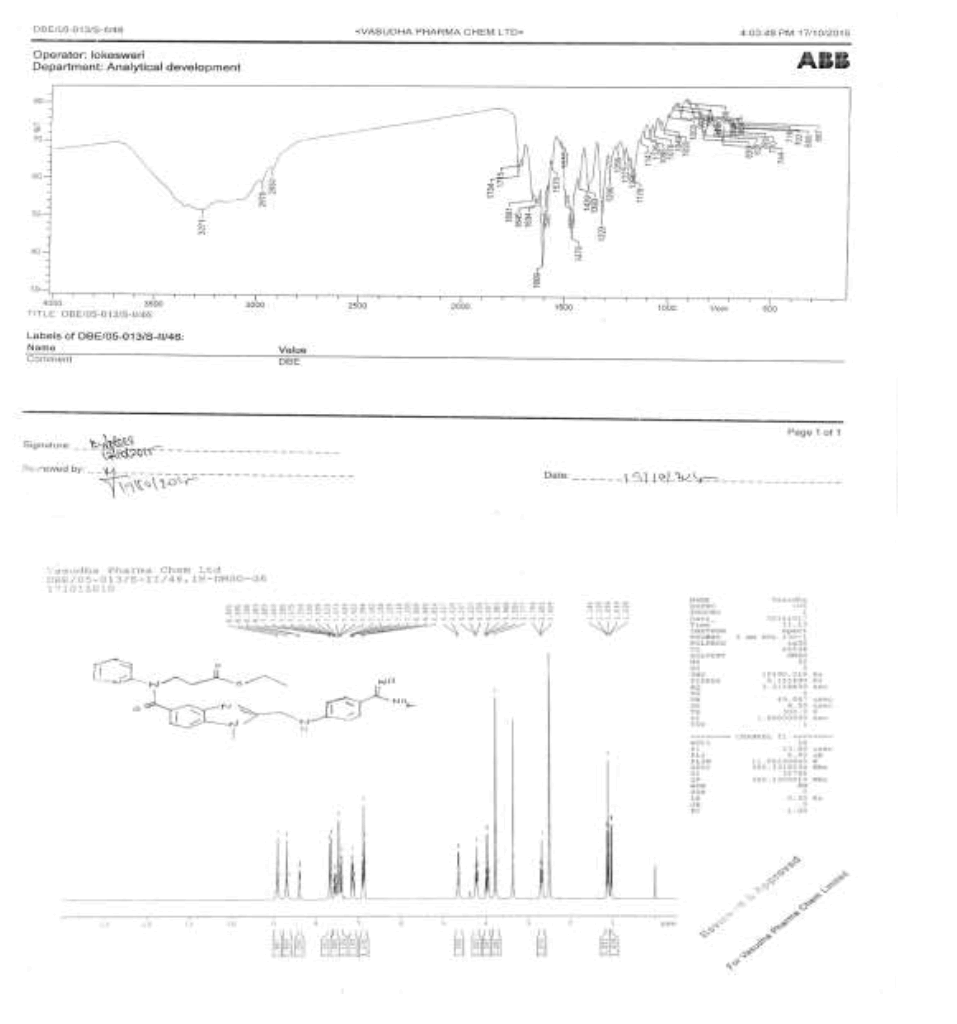
To a mixture of N-[[2-[[[4-[[(Hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5-yl]carbonyl]-N-2- pyridinyl-β-alanine ethyl ester methane sulfonate (50.0 g, 0.069 mol, VI), and water (500.0 ml), the reaction mixture stirred for 48 h at 60- 65°C. Monitor reaction by TLC. After completion of reaction, isolate the solid by filtration. To the crude solid in methanol (200.0 ml) and the reaction mixture was heated to 60-65°C and stirred for 30 min. Cool the reaction mass to 20-25°C and stirred for 2 h. Isolate the solid filtrate and washed with methanol (50.0 ml) and dried to the constant weight below 50°C under reduced pressure to yield ethyl-3-{[(2-{[(4- {[({hexyloxy}carbonyl)amino]carbonyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2-yl)amino}propanoate (IMP-C, Yield 42.5 g, 98%).
IR (cm-1): 3346 (N-H stretching), 2957 (-C-H stretching in -Ar), 1749 (-C=O stretching in ester), 1736 (-C=O stretching in amide), 1601-1647 (- C=N stretching), 1286 (-C-O stretching in ester); 1H-NMR (CDCl3, δ ppm): 10.44 (s, 1H), 8.38 (d, 1H), 7.68 (d, 2H), 7.57-7.51 (m, 1H), 7.47 (s, 1H), 7.39 (d, 1H), 7.17-7.05 (m, 3H), 6.88 (d, 1H), 6.74 (d, 2H), 4.59 (d, 2H), 4.22 (t, 2H), 4.08 (t, 2H), 4.0-3.9 (m, 2H), 3.76 (s, 3H), 2.67 (t, 2H), 1.65-1.56 (m, 2H), 1.37-1.27 (m, 6H), 1.11 (t, 3H), 0.86 (t, 3H); 13C-NMR (CDCl3, δ ppm): 171.0, 170.31, 164.82, 156.0, 153.6, 151.9, 148.62, 140.8, 137.86, 137.23, 130.20, 129.30, 122.79, 122.09, 121.22, 120.13, 119.50, 111.22, 109.46, 64.64, 59.99, 44.34, 33.0, 30.88, 29.89, 28.24, 24.94, 22.0, 13.95-13.87; Mass: 628.70 (M+).
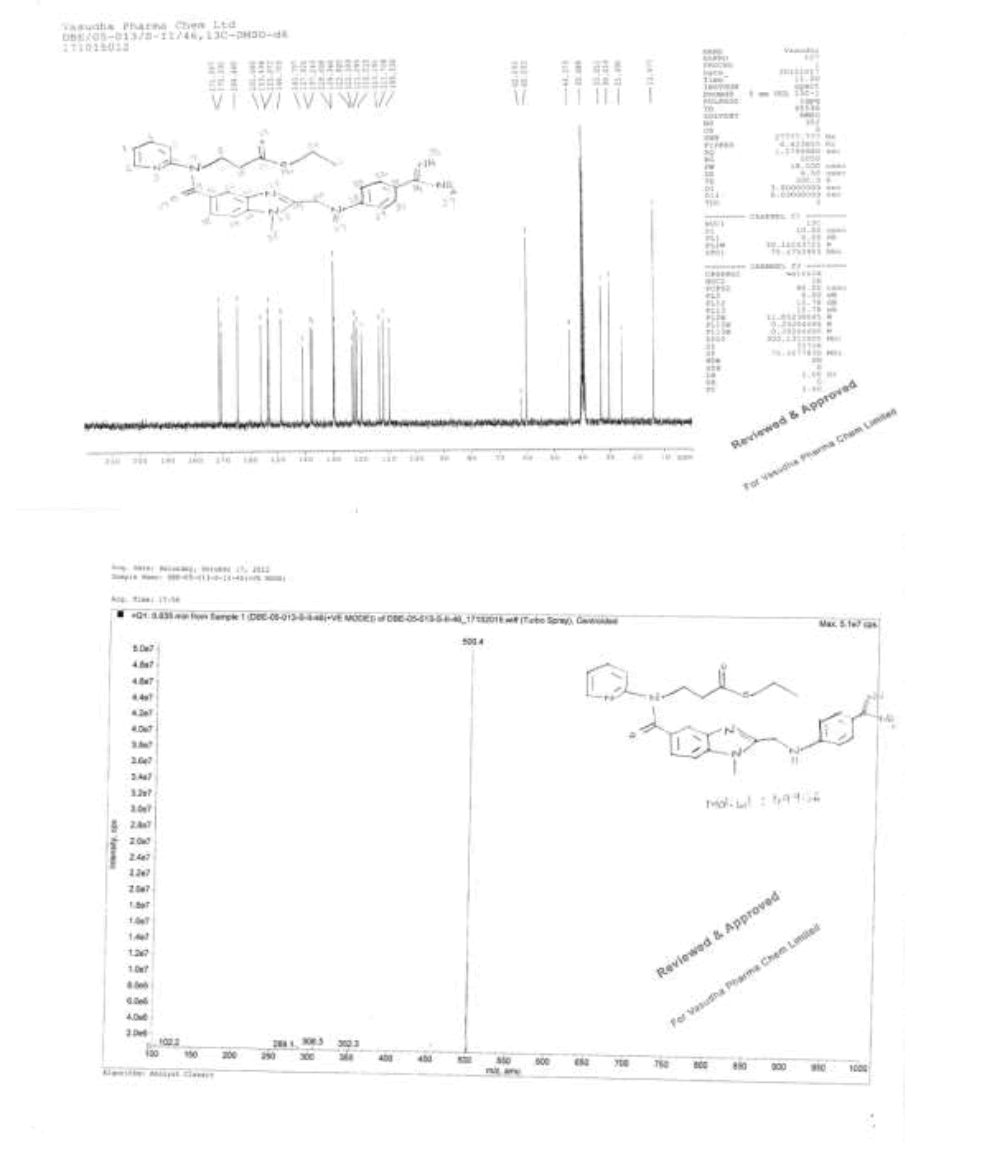
Synthesis of methyl-3-{[(2-{[(4-{[(hexyloxy)carbonyl]carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5- yl)carbonyl](pyridin-2-yl)amino}propanoate (Impurity D):
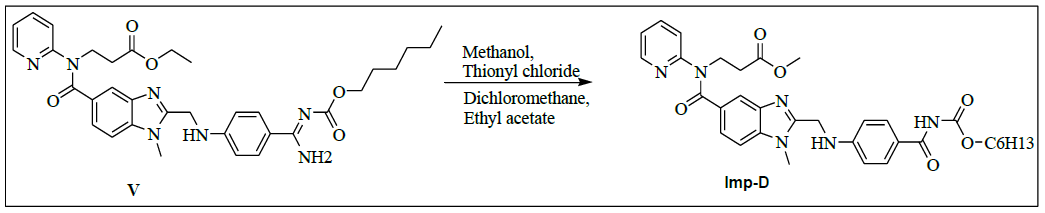
To a mixture of ethyl-3-[[[2-[[[[[[(hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5- yl]carbonyl](pyridin-2-yl)amino]propanoate (50.0 g, 0.079 mol, V) and ethanol (500.0 mL), 10% sodium hydroxide solution (50.0 ml) added, then reaction mixture stirred for 2 h. Monitor the reaction by TLC, once after completion distill off ethanol completely under reduced pressure below 50°C. To the reaction mixture water added and adjusted pH to 4-5 with 10% Con. Hydrochloric acid (21.2 ml) and stir the reaction mass for 1 h at 25-30°C. Filtered reaction mass, washed with water (200.0 mL), and dried to the constant weight below 50°C under reduced pressure to yield 3-{[(2-{[(4-{[(hexyloxy)carbonyl] carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2- yl)amino}propanoicacid (IMP-A, Yield 43.0 g, 90%).
3-{[(2-{[(4-{[(Hexyloxy)carbonyl]carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazo-1-5-yl)carbonyl](pyridin-2- yl)amino}propanoic acid (50.0 g, 0.083 mol, IMP-A) in methanol (350.0 ml) is stirred for 30 min. Thionyl chloride (19.8 g, 0.166 mol) added to the reaction mixture below 20 °C. Stir the reaction mixture for 2 h at 25-30°C. Monitor reaction by TLC. After completion of the reaction distill off methanol and thionyl chloride under reduced pressure below 50°C. Water (300.0 ml) added to the reaction mixture and adjusted pH to 8-9 with 10% sodium carbonate solution (45.0 ml) and extracted with dichloromethane (300.0 ml). Distill off dichloromethane completely the organic layer under reduced pressure below 50°C to get the residue. Ethyl acetate (350.0 ml) added to the residue and stirred for 30 min. Isolated the solid and washed with ethyl acetate (100.0 ml) and dried to the constant weight below 50°C under reduced pressure to yield methyl-3-{[(2- {[(4-{[(hexyloxy)carbonyl]carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl) carbonyl](pyridin-2-yl) amino}propanoate (IMP-D, Yield 48.0 g, 94%); IR (cm-1): 3346 (-N-H stretching), 3065 (-C-H stretching in -Ar), 1736 9(-C=O stretching in ester), 1609-1610 (- C=N stretching), 1572 (-C=C stretching in -Ar), (-C-O stretching in ester).
1H-NMR (CDCl3, δ ppm): 8.9 (s, 2H), 8.38 (d, 1H), 7.79 (d, 2H), 7.54 (t, 1H), 7.473 (s, 1H), 7.39 (d, 1H), 7.17-7.09 (m, 2H), 6.978 (t, 1H), 6.88 (d, 1H), 6.75 (d, 2H), 4.59 (d, 2H), 4.22 (t, 2H), 3.96 (t, 2H), 3.767 (s, 3H), 3.521 (s, 3H), 2.69 (t, 2H), 1.59-1.55 (m, 2H), 1.28 (s, 6H), 0.86 (t, 3H); 13C-NMR (CDCl3, δ ppm): 171.55, 170.41, 166.47, 164.29, 156.03, 153.76, 151.61, 148.73, 140.86, 137.93, 137.29, 129.34, 129.22, 122.85, 122.14, 121.14, 121.30, 121.09, 119.55, 111.43, 109.51, 64.15, 51.44, 44.42, 39.95, 32.83, 31.06, 29.95, 28.58, 25.28, 22.10, 13.94; Mass: 614.4 (M+).
Synthesis of ethyl-3-(2-((4-cyanophenylamino)methyl)-1-methyl-N-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamido)propanoate oxalate (III):
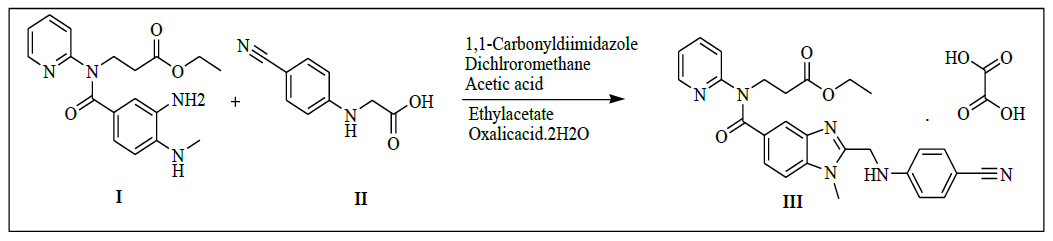
To a mixture of 2-(4-cyanophenylamino)acetic acid (61.6 g, 0.350 mol, II) and dichloromethane (600.0 ml), 1,1-carbonylimidiazole (66.3 g, 0.409 mol) added slowly and stirred for 30 min. Ethyl-3-((3-amino-4-(methylamino)benzoyl)(pyridine-2-yl)amino)propanoate (100.0 g, 0.292 mol, I) in dichloromethane (300.0 ml) was added slowly to the reaction mass and stirred for 8 h. Monitored the reaction by TLC. After completion of the reaction, water (500.0 ml) added and stirred for 30 min. Both organic and aqueous layers are separated. The organic layer is washed with water (500.0 ml) and concentrated under reduced pressure below 40°C to get the residue. To the residue ethyl acetate (600.0 ml) and Acetic acid (100.0 ml) are added and reaction mixture was heated to 80-85°C, stirred for 2 h. To the reaction mass, Water (600.0 ml) is added and stirred for 30 min. Both organic and aqueous layers are separated. Then organic layer washed with 5% sodium bicarbonate solution (200.0 ml). Separated the organic layer and added oxalic acid dihydrate (35.0 g, 0.277 mol). Then reaction mixture stirred for 1 h at 25 to 30°C. Isolated the solid, washed with ethyl acetate (100.0 ml) and dried to a constant weight at 60-65°C to yield ethyl-3-(2-((4- cyanophenylamino)methyl)-1-methyl-N-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamido)propanoate oxalate (III, yield 134.4 g, 80.5%).
IR (cm-1): 3273 (-N-H stretching), 2939 (-C-H stretching in -Ar), 1728 (-C=O stretching in amide), 1753 (-C=O stretching in ester), 1568 (-C=C stretching in -Ar); 1H-NMR (CDCl3, δ ppm): 13.90 (s, 1H), 8.38 (d, 1H), 7.55 (t, 1H), 7.52-7.40 (m, 4H), 7.281 (s, 1H), 7.18-7.09 (m, 2H), 6.89 (d, 1H), 6.81 (d, 2H), 4.60 (s, 2H), 4.22 (t, 2H), 4.08-3.93 (m, 2H), 3.76 (s, 3H), 2.68 (t, 2H), 1.11 (t, 3H); 13C-NMR (CDCl3, δ ppm): 171.08, 170.27, 161.26, 156.0, 153.42, 151.75, 148.74, 140.15, 137.95, 137.07, 133.38, 129.68, 123.09, 122.15, 121.32, 120.45, 119.29, 112.45, 109.71, 96.85, 60.06, 44.43, 39.65, 33.07, 30.0, 13.99.
Mass: 483.4 (M+)-Free Base.
Synthesis of ethyl-l 3-(2-((4-amidinophenylamino)methyl)-1-methyl-N-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamido)propanoate hydrochloride (IV):
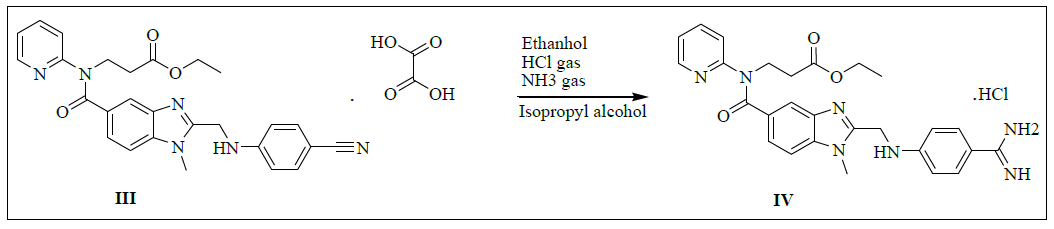
To a mixture of 3-(2-((4-cyanophenylamino)methyl)-1-methyl-N-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamido)propanoate oxalate (100.0 g, 0.174 mol, III) and Ethanol (1000.0 ml), dry HCl gas was added slowly passed to the get 35-40% and stirred for 8 h for reaction completion. Monitor the completion of reaction by TLC and concentrated under reduced pressure below 40°C to get the residue. To the residue, ethanol (1000.0 ml) was added and adjusts pH: 9-10 by using dry ammonia gas and stirred for 10 h for reaction completion. Distill off ethanol completely under reduced pressure at below 50°C to get the residue. To the residue isopropyl alcohol (1000.0 ml) was added and stirred for 30 min. Isolate the solid filtrate and washed with isopropyl alcohol 1200.0 ml) and dried to a constant weight at 65-70°C to yield ethyl-3-(2-((4- amidinophenylamino)methyl)-1-methyl-N-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamido)propanoate hydrochloride (IV, yield 80.4 g, 86.4%); IR (cm-1): 3271 (-N-H stretching), 2978 (-C-H stretching in -Ar) 1634-1645 (-C=N stretching), 1730 (-C=O stretching), 1587 (-C=C stretching in -Ar); 1H-NMR (CDCl3, δ ppm): 8.90 (s, 2H), 8.69 (s, 2H), 8.38 (d, 1H), 7.66 (d, 2H), 7.55(t, 1H), 7.47-7.39 (m, 3H), 7.16 (t, 2H), 6.87 (t, 3H), 4.64 (d, 2H), 4.22 (t, 2H), 4.0 (t, 2H), 3.77 (s, 3H), 2.67 (t, 2H), 1.11 (t, 3H); 13C-NMR (CDCl3, δ ppm): 171.05, 170.33, 164.44, 155.99, 153.43-153.07, 148.70, 140.79, 137.92-153.07, 148.0, 140.79, 137.92-137.24, 129.65-129.36, 122.81-122.10, 121.29, 119.51, 113.19, 111.70, 109.53, 60.03, 44.37, 33.05, 30.0, 25.49, 13.97; Mass: 536.03 (M+).
Synthesis of N-[[4-[[[5-[[(3-Amino-3-oxopropyl)-2-pyridinylamino]carbonyl]-1-methyl-1H-benzimidazol-2- yl]methyl]amino]phenyl]iminomethyl]-carbamic acid hexyl ester (Impurity G):
To a mixture of ethyl-3-[[[2-[[[[[[(hexyloxy)carbonyl]amino]iminomethyl]phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5- yl]carbonyl](pyridin-2-yl)amino]propanoate (50.0 g, 0.079 mol, V) and ethanol (500.0 ml), 10% sodium hydroxide solution (50.0 ml) added, then reaction mixture stirred for 2 h. Monitor the reaction by TLC, once after completion distill off ethanol completely under reduced pressure below 50°C. To the reaction mixture water added and adjusted pH to 4-5 with 10% Con. Hydrochloric acid (21.2 ml) and stirs the reaction mass for 1 h at 25-30°C. Filtered reaction mass, washed with water (200.0 mL), and dried to the constant weight below 50°C under reduced pressure to yield 3-{[(2-{[(4-{[(hexyloxy)carbonyl] carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazol-5-yl)carbonyl](pyridin-2-yl) amino}propanoic acid (IMP-A, Yield 43.0 g, 90%).
To a mixture 3-{[(2-{[(4-{[(hexyloxy)carbonyl]carbamimidoyl}phenyl)amino]methyl}-1-methyl-1H-benzimidazo-1-5-yl)carbonyl](pyridin-2- yl)amino}propanoicacid (50.0 g, 0.083 mol, IMP-A), Dimethylformamide (50.0 ml), 1,1-dicarbonyldiimidazole (13.4 g, 0.083 mol) and stirred for 30 min at 25-30°C. Cool the reaction mass to 10°C and adjust pH: 9-10 by using dry ammonia gas and stirred for 1 h at 25-30°C. Monitor the completion of reaction by TLC. Water (300.0 ml), was added to the reaction mixture and extract with Ethyl acetate (250.0 ml). Stirred the reaction mixture and separate the organic layer and aqueous layer. Washed the organic layer with saturated brine solution (100.0 ml). Separate the organic layer and concentrated the organic layer under reducing pressure at below 50°C. To the residue, cyclohexane (200.0 ml) was added and stirred for 4 h at 25 to 30°C. Isolate the solid filtrate and washed with cyclohexane (100.0 ml) and dried to the constant weight below 50°C under reduced pressure to yield N-[[4-[[[5-[[(3-Amino-3-oxopropyl)-2-pyridinylamino] carbonyl]-1-methyl-1H-benzimidazol-2-yl] methyl] amino] phenyl]iminomethyl]carbamic acid hexyl ester (IMP-G, Yield 40.9 g, 82%).
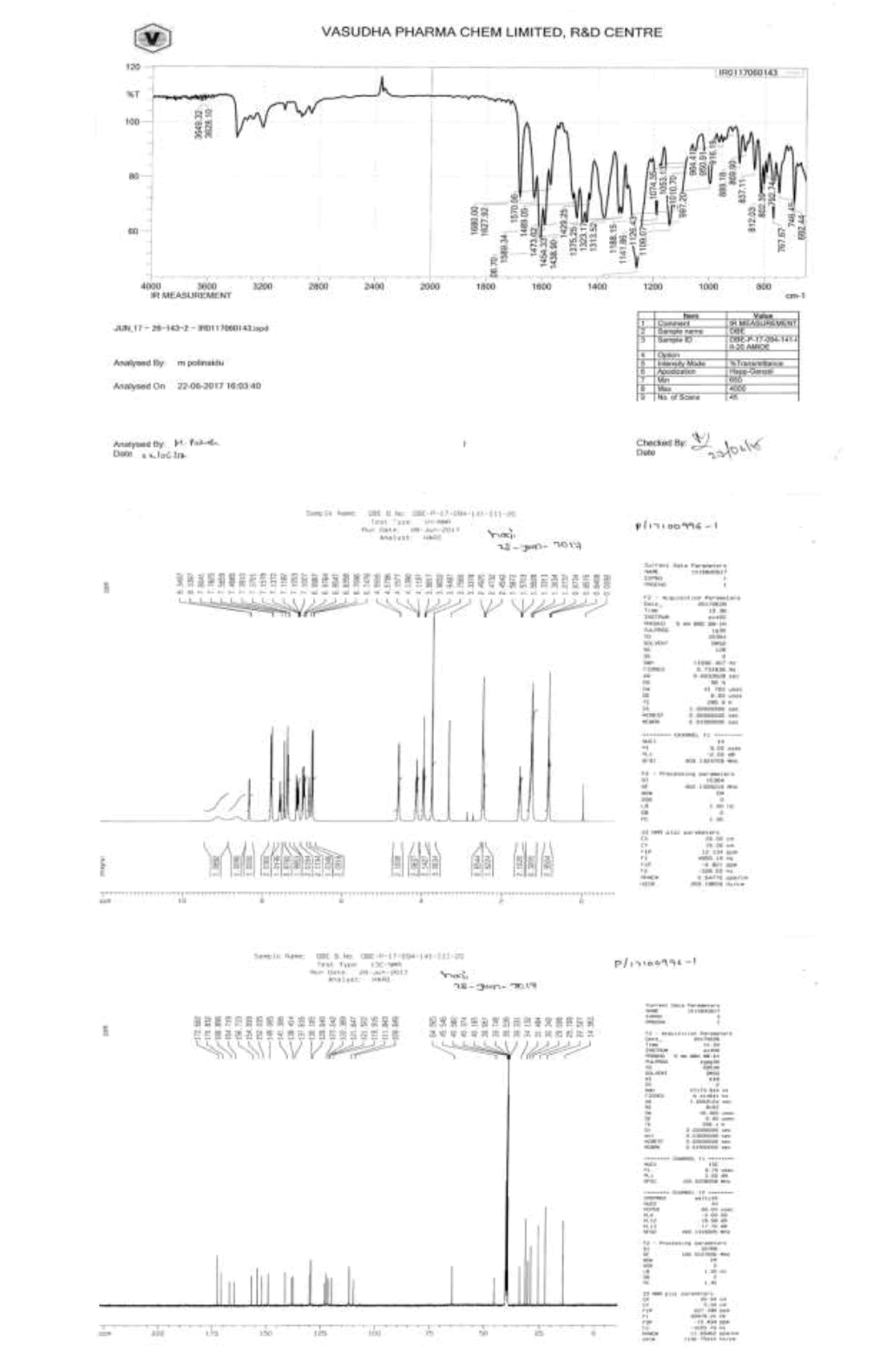
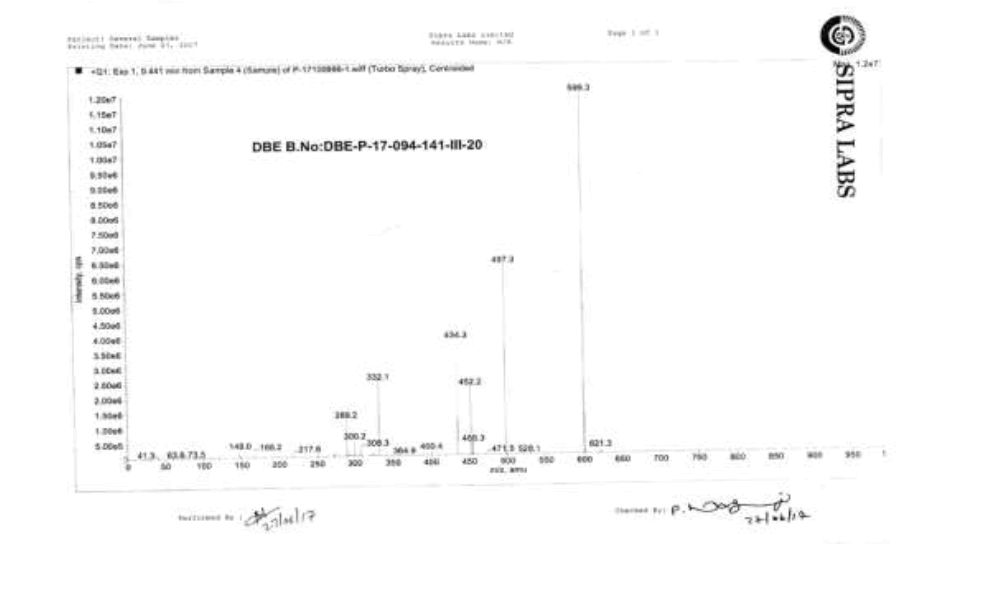
IR (cm-1): 3649.32 (-N-H stretching 1°), 3628.10 (-N-H stretching 2°), 1680.0 (-C=O stretching in ester), 1627.92 (-C=O stretching in amide), 1606.70 (-C=N stretching), 1589.34 (-C=C- stretching in -Ar), 1570.06 (-C=O stretching), 1489.05 (-C-O stretching in ester); 1H-NMR (CDCl3, δ ppm): 8.50-9.50 (Hump, 2H), 8.34 (d, 1H), 7.79 (d, 2H), 7.56 (t, 1H), 7.46 (s, 1H), 7.38 (d, 2H), 7.15-7.08 (m, 2H), 6.99-6.94 (m, 2H), 6.83 (s, 1H), 6.75 (d, 2H), 4.58 (d, 2H), 4.13 (t, 2H), 3.96 (t, 2H), 3.75 (d, 3H), 2.47 (t, 2H), 1.56 (q, 2H), 1.30 (s, 6H), 0.85 (t, 3H); 13C-NMR (CDCl3, δ ppm): 172.68, 170.83, 166.89, 164.71, 156.73, 154.09, 152.03, 149.08, 141.30, 138.41, 137.61, 130.10, 129.64, 123.24, 122.38, 121.64, 121.52, 119.91, 109.84, 64.56, 64.33, 45.54, 40.58, 34.13, 31.48, 30.34, 29.00, 25.70, 22.52, 14.36; Mass: 598.7 (M+).
Impurity-A
Impurity B
Impurity C
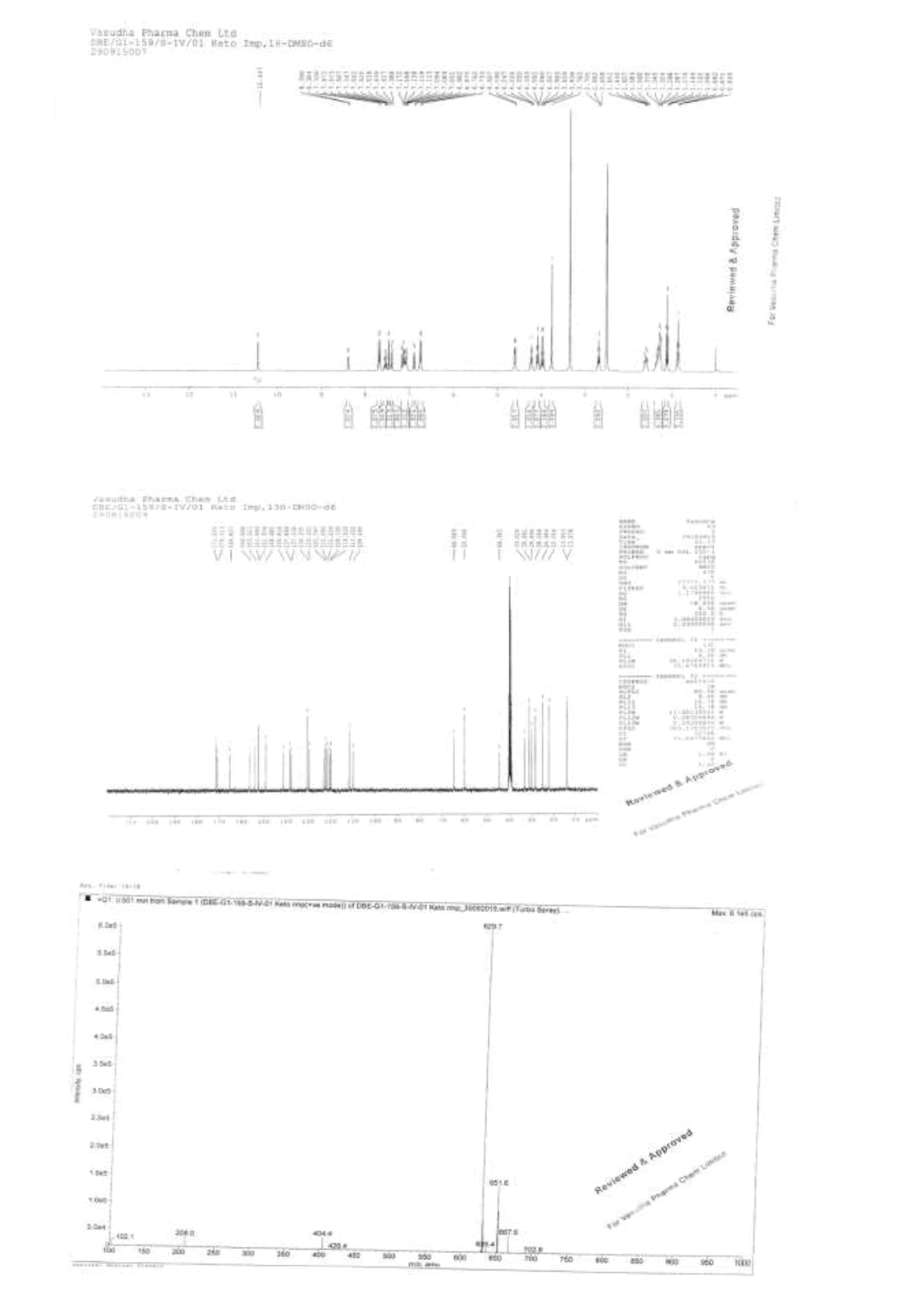
Impurity D
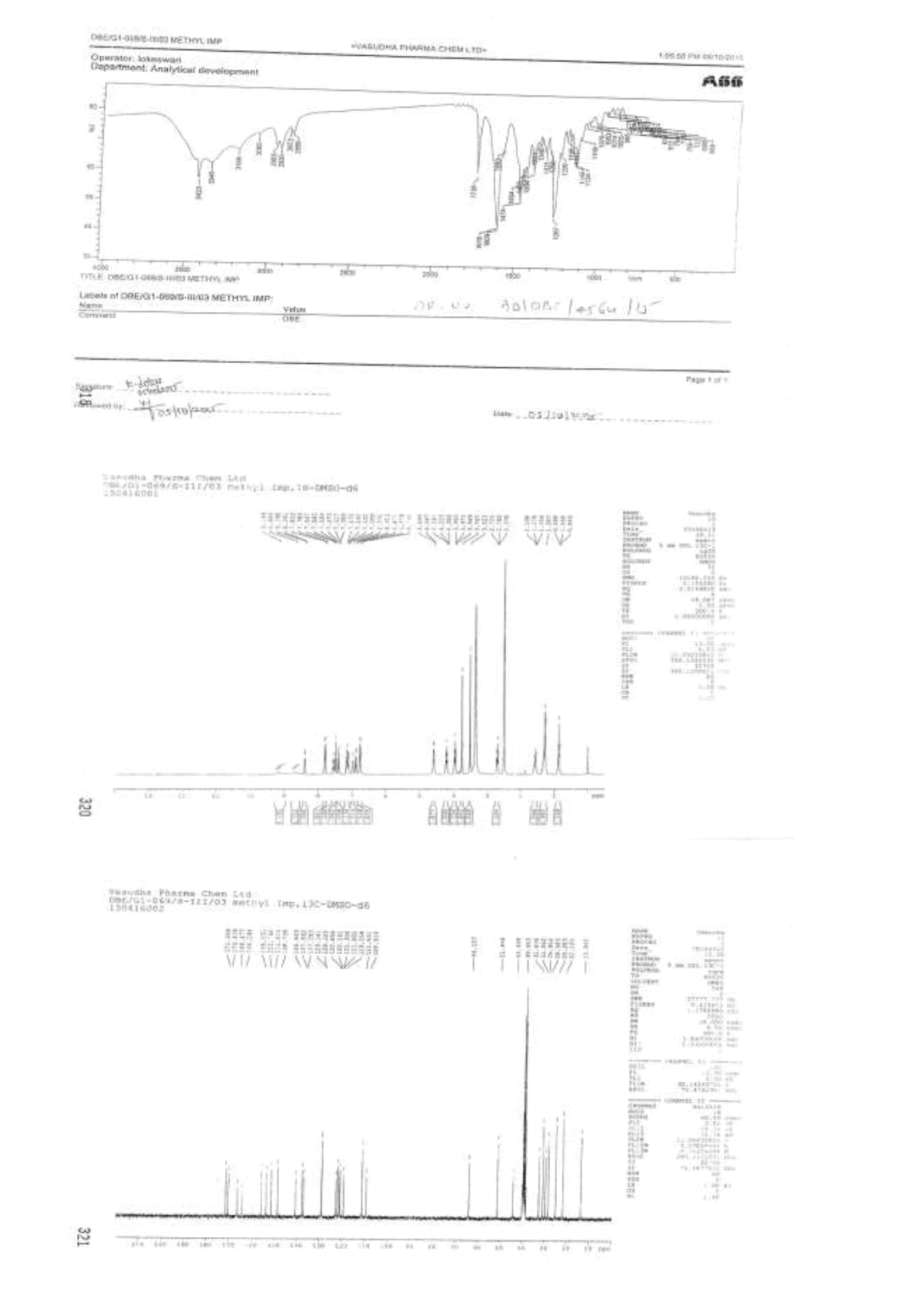
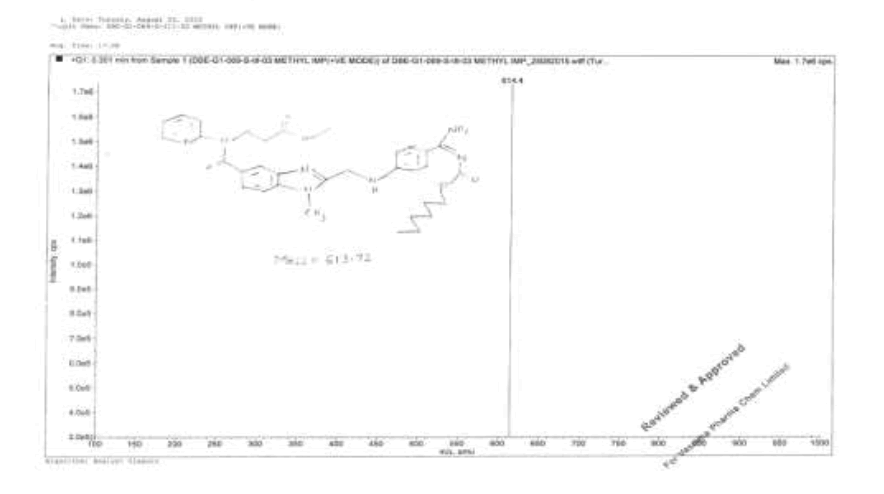
Impurity E
Impurity F
Impurity G