Review Article - Der Pharma Chemica ( 2019) Volume 11, Issue 3
An Insight into Progress of Organic Motif Anti HIV Drug Discovery
Atukuri Dorababu*Atukuri Dorababu, Department of Chemistry, SRMPP Govt. First Grade College, Huvinahadagali-583219, Karnataka, India, Email: dora1687@gmail.com
Abstract
HIV, a dreadful virus has infected billions of people worldwide. It is spreading like anything despite the advances in the drug discovery. Even though innumerable strategies were designed most of them were in vain as the HIV virus has got inbuilt genetic mutation capability. Alongside, the virus is getting resistance to the chemical drugs. The chemical drugs are synthesized so as to target a particular protein of HIV which is most essential for its life. Few classes of drugs target the viruses in their latent stage and thereby reducing viral load. Some important classes of chemical drugs entail diarylpyrimidines, methoxy indoles and methoxy azoindoles etc. Some of the natural products are also derivatized to afford potent anti-HIV drugs. FDA (Food and Drug Administration) has approved a large number of drugs after clinical investigation under different phases. According to mode of their action, they have been classified as antifibrotics, CCR5 antagonists, integrase inhibitors, latency reversing agents, maturation inhibitors, (NNRTIs), (NRTIs), protease inhibitors, rev inhibitors, etc. Here in we reported the chemicals drugs synthesized from the past to present which includes a wide variety of synthetic chemical drugs possessing anti HIV potency. Also, the various drugs approved by the FDA were described.
Keywords
Anti HIV drugs, CD4-T cells, HIV, Immune system, Retrovirus
Introduction
A disease that makes a person go trembling, despair, dread, terror and blunder is HIV infection which stands for Human Immunodeficiency Virus. It is a lentivirus that causes the HIV infection at the initial stage and leads to Acquired Immuno Deficiency Syndrome (AIDS) over time untreated. AIDS is a condition in which successive break down of immune system occurs making room for the opportunistic infections, cancer and other diseases in the body. HIV suppresses immune system by infecting crucial cells in the immune system such as helper T cells CD4+ T cells, macrophages and dendritic cells. Infected CD4+ T cells are killed through various mechanisms and there by abatement of CD4+ T cell count in the human body. Decline of CD4+ T cell count below a critical level leads to complete deterioration of cell-mediated immunity. Hence body will progressively allow the opportunistic diseases to infect in the body progressing to the development of the condition AIDS. The structure of HIV particle is as depicted in the Figure 1.
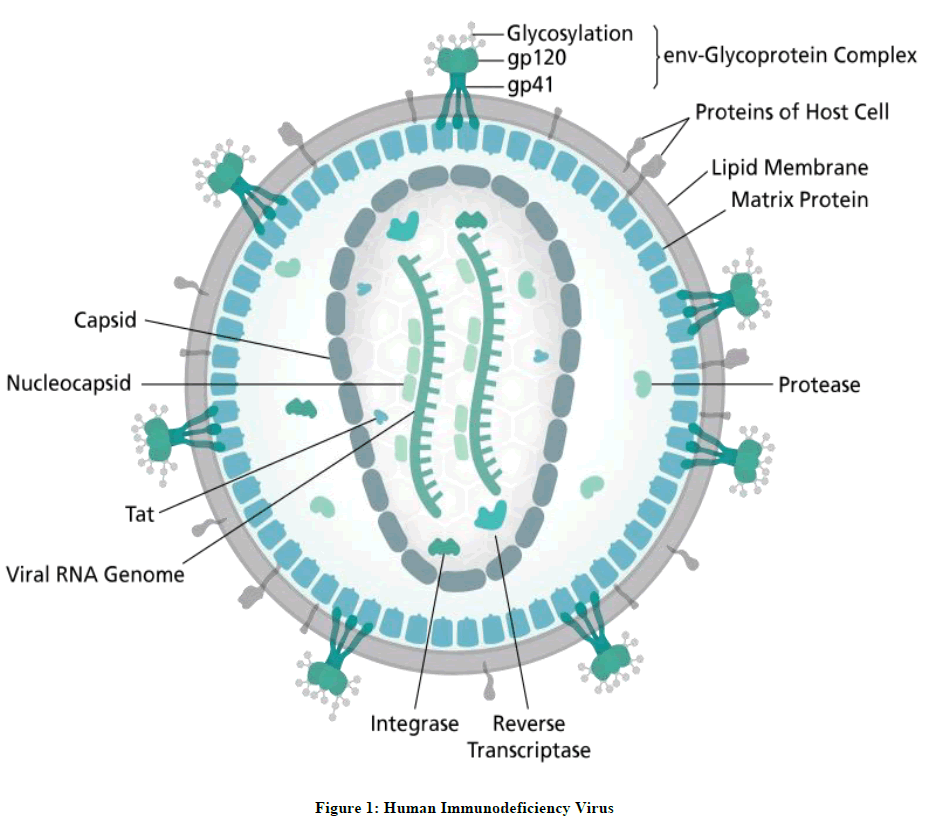
Figure 1: Human Immunodeficiency Virus
Transmission of HIV occurs mainly through sexual contact, blood transfusion from HIV infected person, infected mother to her infant during pregnancy, child birth by exposure to her blood or vaginal fluid and though breast milk. Advance HIV infection treatment involves prolonged prescription of Anti-Retro Viral drugs (ARV) to the infected person.
These drugs are always given with other ARVs; this combination therapy of ARVs is known as Anti-Retroviral Therapy (ART). It involves taking appropriate ART that slow down the progression of the virus in the body. The ART deteriorates the viral load in the body and body fluids and prolongs the life of a person by reducing the risk of opportunistic diseases. Also it reduces the chance of transmission to other person. Before development of anti-retro viral drugs, a lot of research has been done for synthesis of anti HIV drugs. Heterocyclic synthesis and pharmacology are the fields where research of HIV prevention, treatment and cure is being done. We have carried out a wide literature survey with respect to origin, progress and development and advancement of anti HIV drugs. In our present work, we have emphasized the diversity in the chemical drugs and chemical scaffolds upon which these chemical drugs were built. Also the mode of action of the different chemical drugs in prevention, cure and prolonging of the patients′ life is discussed. Correlation of the pharmacological activity of the synthesized drugs with those of standard HIV drugs is also attempted.
Progress of research in the HIV drug discovery
3-Fluoro-2-(phosphonomethoxy)propyl (FPMP) derivatives
Nucleoside derivatives of acyclic nucleoside phosphonates (ANPs) were have reported to have excellent antiviral activities especially against DNA viruses and retroviruses [1]. Among ANPs derivatives, 3-Fluoro-2-(phosphonomethoxy) propyl (FPMP) analogues characterized by the presence of fluorine atom in the acyclic side chain which have shown moderate activity against HIV and HBV [2,3].
Min Luo et. al. [4] have synthesized a set of molecules (nucleoside derivatives of ANPs, Figure 2) which were screened for antiviral activity by luciferase assay in TZM-bl cells against the HIV strains, CXCR4-tropic HIV-1 strain and CCR5-tropic HIV-1 strain. Tenofovir, tenofovir disoproxil fumarate and tenofovir alafenamide were used as standard drugs. Among the tested molecules, annoyingly, compounds 1a/b, 2a/b, 3a/b have not exhibited ant-HIV activity (IC50>100 μM). Whereas the compounds 4 (IC50=4.80 μM) and 5 (IC50=1.81 μM) have shown a significant anti-HIV activity which indicated that the 2-aminosuccinate appendant has increased lipophilicity and hence cellular permeability is improved.
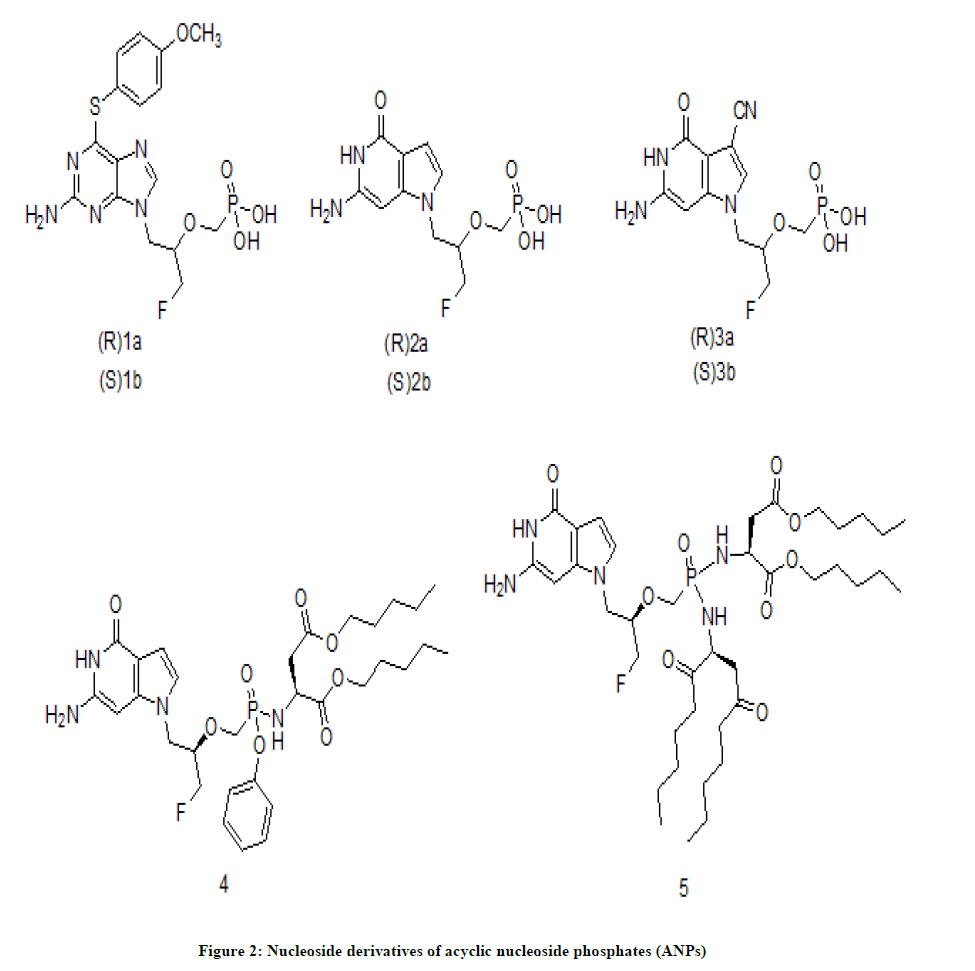
Figure 2: Nucleoside derivatives of acyclic nucleoside phosphates (ANPs)
Betulinic acid derivatives
A series of Betulinic acid derivatives [5] were prepared by making modification to the maturation inhibitor, GSK3532795. The modification was made at C-17 with a variety of moieties in one of the schemes (Figure 3). The synthesized compounds were evaluated for their antiviral activity against WT HIV-1, V370A, and ΔV370 polymorphic viruses in a virus replication assay. Except a few, all compounds exhibited high metabolic stability in vitro with t1/2. The test compounds were also tested for their in vivo pharmacokinetic activity.
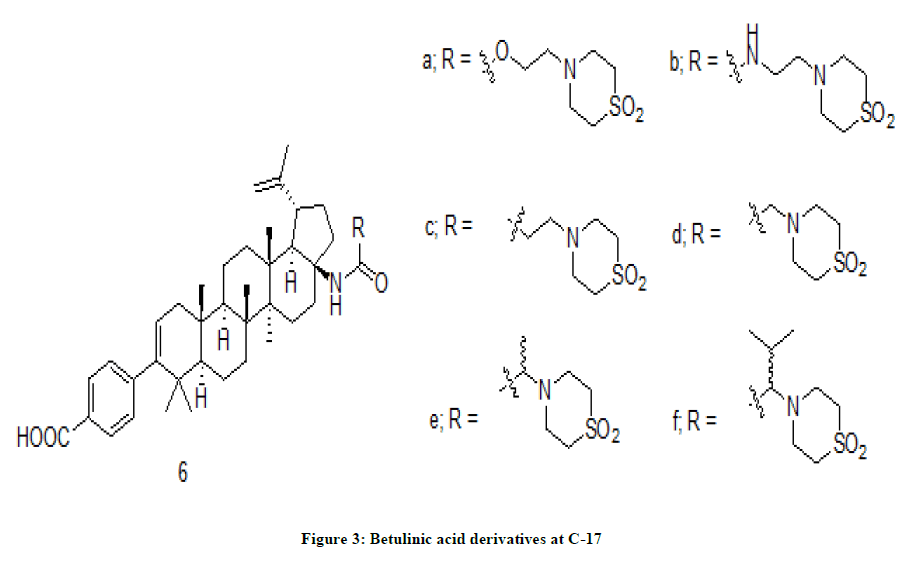
Figure 3: Betulinic acid derivatives at C-17
Diarylpyrimidine prodrugs
Prodrug strategy is one of the main streams in the drug discovery and in synthesis of antivirals [6-8]. These prodrugs are converted to parent drugs by enzymatic metabolism. In this context researchers are have been trying to prodrugs by appending various organic motifs. Here, antiviral drug RDEA427 7, (Figure 4) diarylpyrimidine (DAPY) derivative and a NNRTI was chosen for the preparation of its prodrug 8 (EC50=0.0055 ± 0.0047). RDEA427 (EC50=0.0029 ± 0.0007) inhibits WT HIV-1 and a wide range of mutant HIV-1 strains.
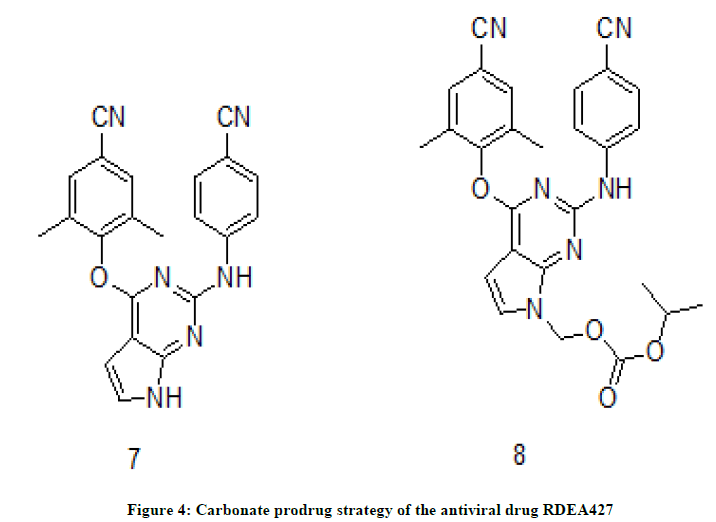
Figure 4: Carbonate prodrug strategy of the antiviral drug RDEA427
The carbonate prodrug and antiviral drug RDEA427 were tested for antiviral activity [2] against HIV-1 WT strain, RES056 and HIV-2 strain, ROD using nevirapine, efavirenz and etravirine as reference compounds. The antiviral activity revealed that the carbonate prodrug exhibited potential antiviral activity against HIV-1. However, its activity is comparably very less than that of the drug REDA427. The prodrug is inactive against the strain HIV-2.
Small molecule macrocyclic HIV-1 Inhibitors
M. Hurevich et. al. have designed CD4 mimetics as gp120 inhibitors and prepared small molecule macrocyclic HIV-1 Inhibitors [9]. They managed to reduce the distance between important pharmacophores such as Phe and Arg on the m side of the backbone of cyclic inhibitor without affecting the antiviral activity. Both synthesized molecules CG-1 11 and SC-1 10 are structural regioisomers which sustain the actual CD4 active pharmacophores but the difference lies in change in the position of functional groups (Figure 5). All the compounds were evaluated for anti-HIV activity in which compound SC-1 has exhibited weak inhibitory activity while the compound CG-1 has inhibited more than 80% of viral infection. The difference in the activity between CG-1 and SG-1 revealed that anti-HIV activity depends not only on the nature for pharmacophores but also upon correct orientation of pharmacologically active groups.
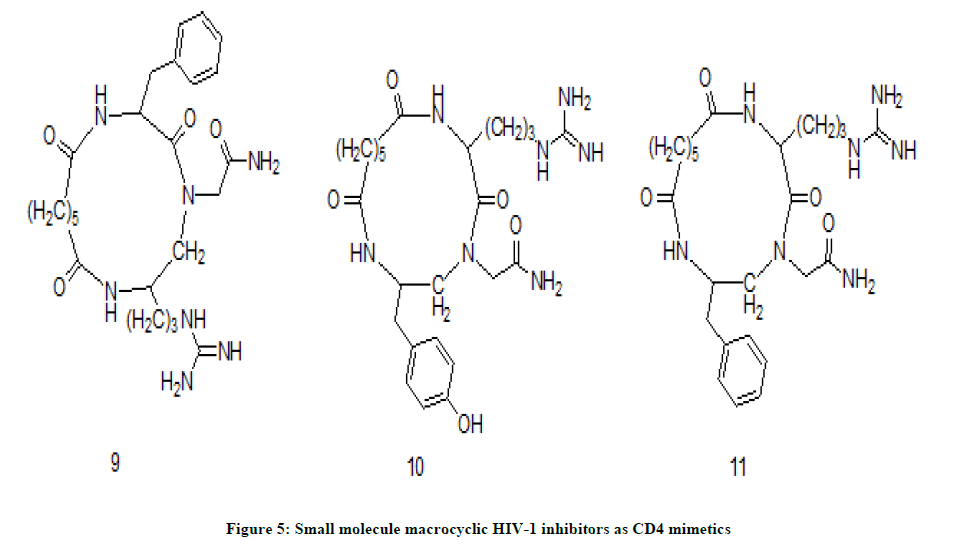
Figure 5: Small molecule macrocyclic HIV-1 inhibitors as CD4 mimetics
C-linked heteroaryl and N-linked heteroaryl C-7 position of 4-methoxy indoles and 4-methoxy 6-azaindoles
In a study T. Wang et. al. [10] have synthesized novel anti-HIV molecules by appending C-linked heteroaryl and N-linked heteroaryl motifs at the C-7 position of 4-methoxy indoles 12 and 4-methoxy 6-azaindoles 13 (Figure 6). Anti-HIV activity revealed that introduction of sp2- hybridized heteroaromatic substituent at the C-7 position of indoles and 6-azaindoles has exhibited potent antiviral activity. An attempt of comparison of anti-HIV activity of C-7 substituted indoles and 6 azaindoles in both cases was made. In the C-7 C-linked derivatives, increased metabolic stability could be correlated to increased polarity of C-7 heteroaryl moiety. Again C-7 N-linked heteroaryl compounds have exhibited higher metabolic stability without significant change of membrane permeability. Data of anti-HIV activity of C-7 nitrogen-linked heteroaryl compounds have shown promising activity compared C-7 carbon- linked heteroaryl compounds.
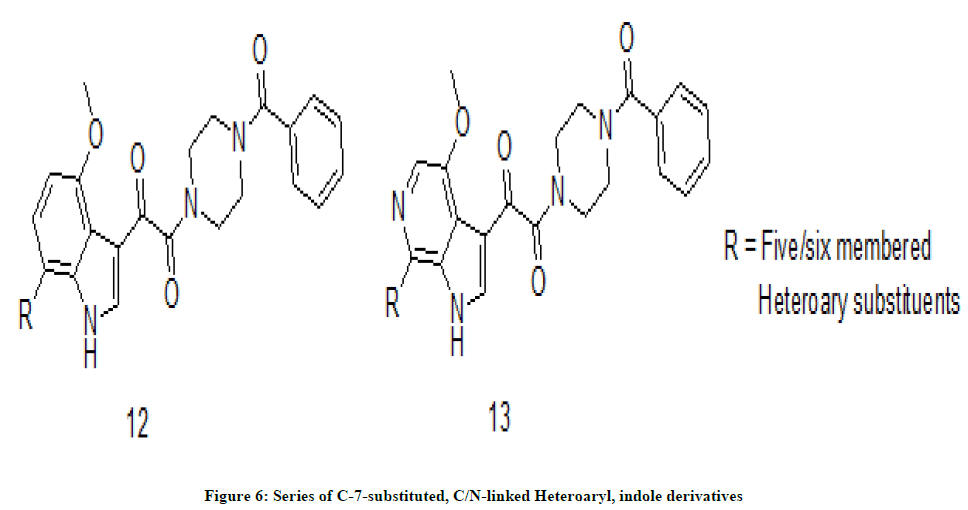
Figure 6: Series of C-7-substituted, C/N-linked Heteroaryl, indole derivatives
A potent anti-HIV drug, attachment inhibitor fostemsavir is converted into its prodrug 14 (Figure 7). Comparison of anti-HIV activity of the prodrug shown that its antiviral potency is 7 fold improved.
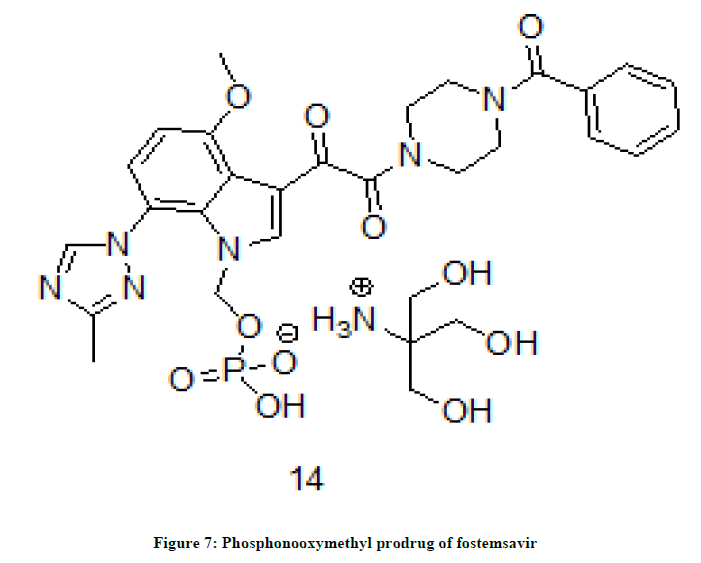
Figure 7: Phosphonooxymethyl prodrug of fostemsavir
Thiophene [3,2-d]pyrimidine derivatives
DAPY based derivatives of NNRTIs were developed as anti-HIV drugs which have exhibited a very potent antiviral activity [11]. A set of thiophene [3,2-d]pyrimidine derivatives with a linker between the thiophenepyrimidine core and right wing [12]. Newly synthesized novel series of hydrazone-substituted thiophene [3,2-d] pyrimidine derivatives were evaluated for anti-HIV activity against WT HIV-1 strain. Among them, four compounds have exhibited striking activity and compound 15 (EC50=21.2 nM) (Figure 8) has shown promising anti HIV activity and lower toxicity.
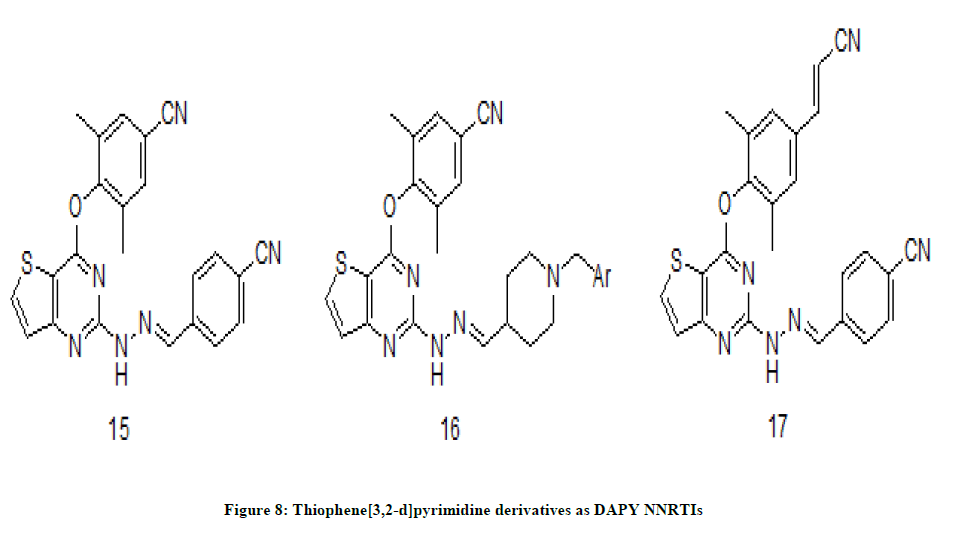
Figure 8: Thiophene[3,2-d]pyrimidine derivatives as DAPY NNRTIs
Aminothiochromane and aminotetrahydronaphthalene carboxamide derivatives
A. K. Ghosh et. al. [13] has designed a series of HIV-1 protease inhibitors by incorporating aminothiochromane and aminotetrahydronaphthalene carboxamide moieties. The design was made to achieve the extensive hydrogen bonding between inhibitors and HIV-1 protease active site. The newly designed HIV-1 protease inhibitors were screened for anti HIV activity in MT-4 human T-lymphoid cells. Anti HIV activity has revealed that the compounds 19 (IC50=47 nM) and 21 (IC50=232 nM) (Figure 9) are potent HIV-1 protease inhibitors among the synthesized compounds. They have also studied that oxidation of ring sulfur of compound 18 (IC50<1000 nM) to sulfone derivative 19 (IC50=47 nM) has increased its pharmacological activity substantially. However, the antiviral activity and enzyme inhibition are reduced when Boc-group of compound 19 was removed to afford compound 20 (IC50<1000 nM). Similar changes in antiviral activity have been observed for other derivatives on oxidation and Boc-group removal.
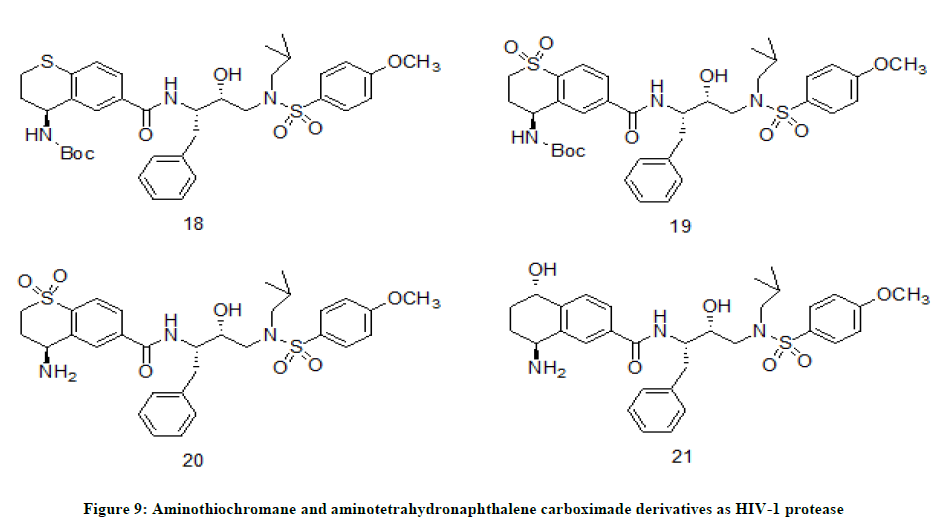
Figure 9: Aminothiochromane and aminotetrahydronaphthalene carboximade derivatives as HIV-1 protease
Sulfonamide derivatives
A new class of HIV-1 protease inhibitors was designed by A.K. Ghosh et. al. [14]. These inhibitors have derivatives of sulfonamide as P2’ ligands and fused tricyclic polyethers as P2 ligands. The ring size of the polyethers and with various substituents was varied and enhancement of interaction of ligand with protease active site. The target molecules are screened for anti HIV activity in MT-2 cells using darunavir and saquinavir as reference compounds. The protease inhibitors 23 (IC50=0.023 nM) and 24 (IC50=0.027 nM) (Figure 10) have 6-5-5 ring system as P2 ligand, aminobenzothiazole as P2’ and difluorophenylmethyl moiety as P1 ligand have shown excellent inhibiting activity in MT-2 cells.
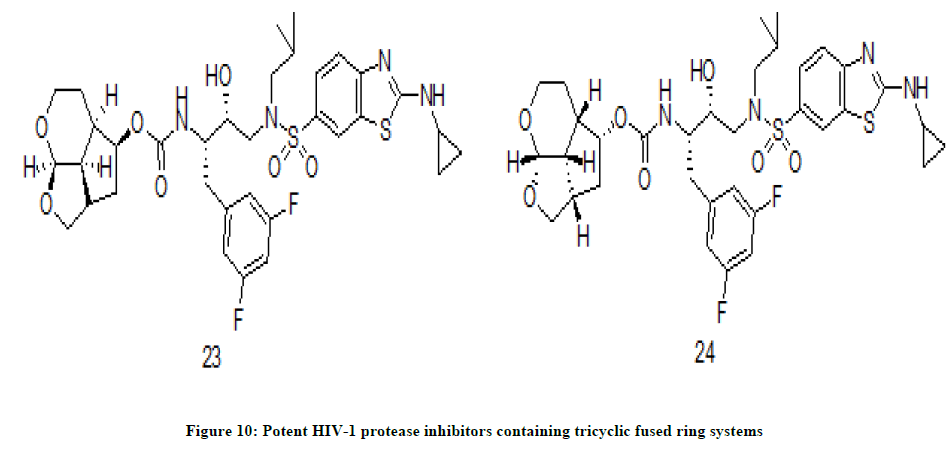
Figure 10: Potent HIV-1 protease inhibitors containing tricyclic fused ring systems
(S)-THF-tertiary amine-acetamide derivatives
One of the best strategies to synthesize effective HIV-1 protease inhibitor and eliminate drug resistance is to enhance extensive hydrogen bond interactions with residues in the active site of the protease enzyme [15,16]. In this context X. Bai et. al. [17] has designed HIV-1 protease inhibitors to optimize the enzymatic activity. In their design, stereochemically defined THF ring and tertiary amine structural template as P2-ligand were incorporated to have potential interactions with HIV-1 protease backbone. All the target molecules were evaluated for their HIV-protease inhibition activity and the results of pharmacological activity were correlated with SAR study. In the pharmacological activity, inhibitors with (S)-THF-tertiary amine-acetamide P2-ligand showed impressive enzymatic activity. The compounds 25 (IC50=0.35 nM), 26 (IC50=0.88 nM) and 27 (IC50=0.77 nM) (Figure 11) exhibited potent inhibitory activity.
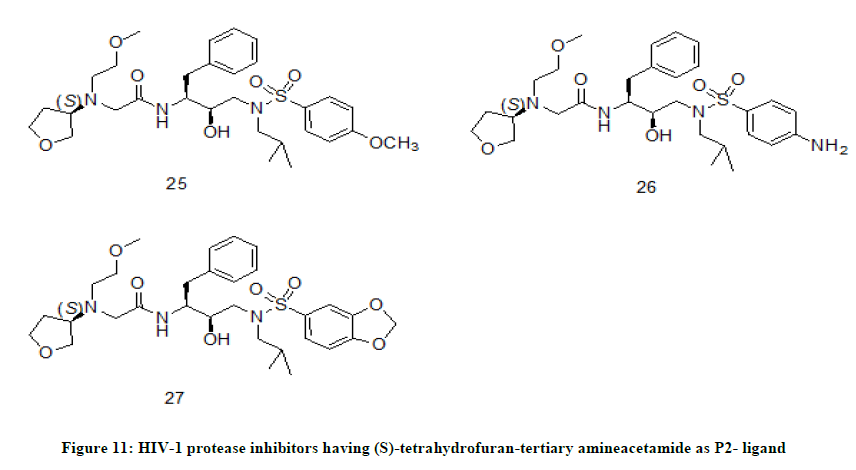
Figure 11: HIV-1 protease inhibitors having (S)-tetrahydrofuran-tertiary amineacetamide as P2- ligand
Oxazolidinone derivatives
In recent years HIV-1 protease inhibitors have become most essential class of chemical drugs for antiretroviral regimen [18,19] as the protease inhibitors inhibit formation of mature viruses. Also, the polypeptide class of chemical drugs has posed a lot problems viz.; poor absorption, metabolic instability etc. [20]. A novel series of potent HIV-1 protease inhibitors were synthesized in which novel bicyclic oxazolidinone derivatives as the P2 ligand utilizing a key o-iodoxybenzoic acid mediated cyclization [21]. The lead compounds were tested for antiviral activity in MT-4 cells using darunavir as a reference compound. In the pharmacological activity compound 28 (IC50=31 nM) and 29 (IC50=41 nM) (Figure 12) have shown antiviral potency against ATV resistant HIV-1 variants. Compound 28 with insertion of N-methylallyl group has resulted in enhanced enzyme inhibitory activity. But reduction of N-methylallyl group to isobutyl group has reduced the activity slightly. However the activity of both compounds 28 and 29 is inferior to the reference compound darunavir (IC50=3.2 nM). They also fail to inhibit the replication of highly darunavir resistant HIV-1 variants.
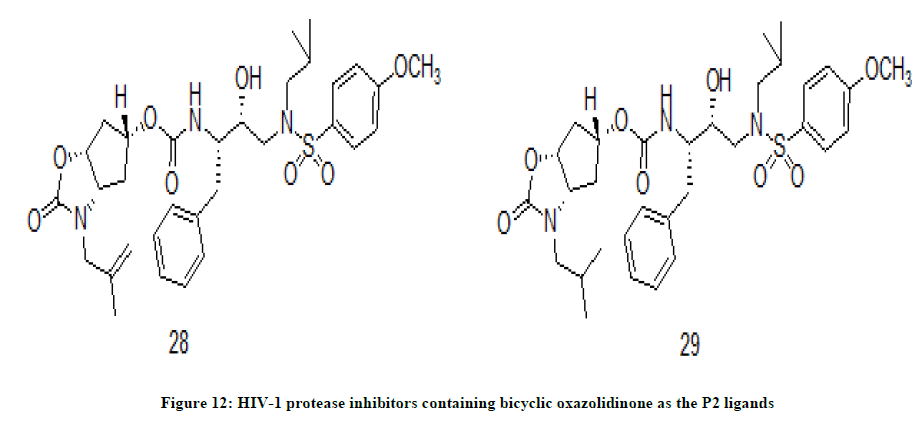
Figure 12: HIV-1 protease inhibitors containing bicyclic oxazolidinone as the P2 ligands
Diaryl ethers derivatives of carboxymethoxyphenacyl moiety
New HIV-1 reverse transcriptase inhibitors developed by T. Fraczec et. al. [22] is designed by appending the diaryl ethers to carboxymethoxyphenacyl scaffold. The resulted compounds are a class of second generation NNRTIs and expected to have higher solubility and chemical stability. Compounds 30 (Figure 13) (IC50=0.36 ± 0.01 nM), 31 (IC50=3.07 ± 0.22 nM), and 32 (IC50=0.65 ± 0.03 nM) were found to be very potent inhibitors of HIV-1 reverse transcriptase enzyme which was comparable to the standard drug NVP (IC50=0.75 ± 0.02 nM).
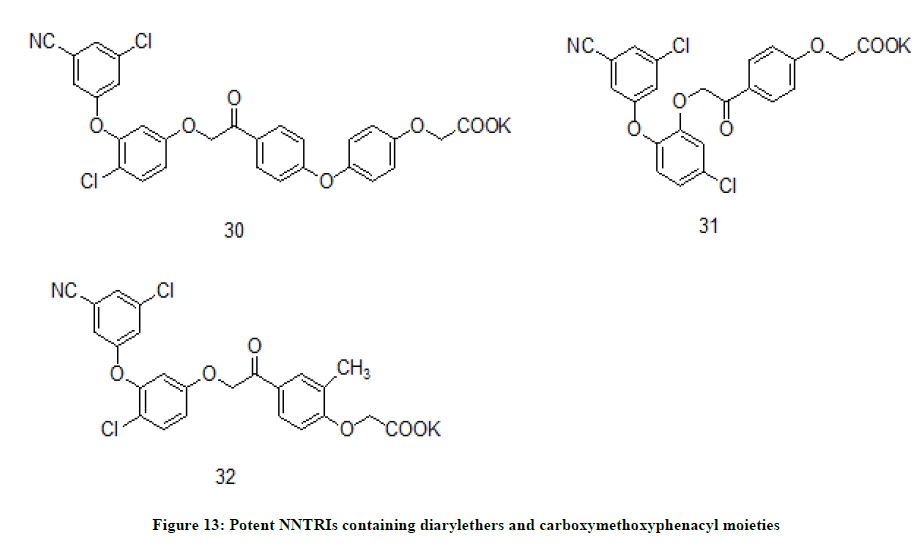
Figure 13: Potent NNTRIs containing diarylethers and carboxymethoxyphenacyl moieties
N1-aryl-2-arylthioacetamido-benzimidazoles and N1-aryl-2-aryloxyacetamido-benzimidazoles
A new set of NNRTIs, N1-aryl-2-arylthioacetamido-benzimidazoles and N1-aryl-2-aryloxyacetamido-benzimidazoles [23] were synthesized and evaluated for HIV-1 reverse transcriptase enzyme inhibition activity in MT-4 cells using Nevirapine and efavirenz as reference compounds. Among N1-aryl-2-arylthioacetamido-benzimidazole derivatives 33 (IC50=0.047 ± 0.007 nM), 34 (IC50=0.05 ± 0.0005 nM) (Figure 14) have shown best inhibition activity and compounds 35 (IC50=0.76 ± 0.19 nM), 36 (IC50=5.84 ± 0.00 nM) (Figure 14) have exhibited excellent inhibition activity among N1-aryl-2-aryloxyacetamido-benzimidazole derivatives.
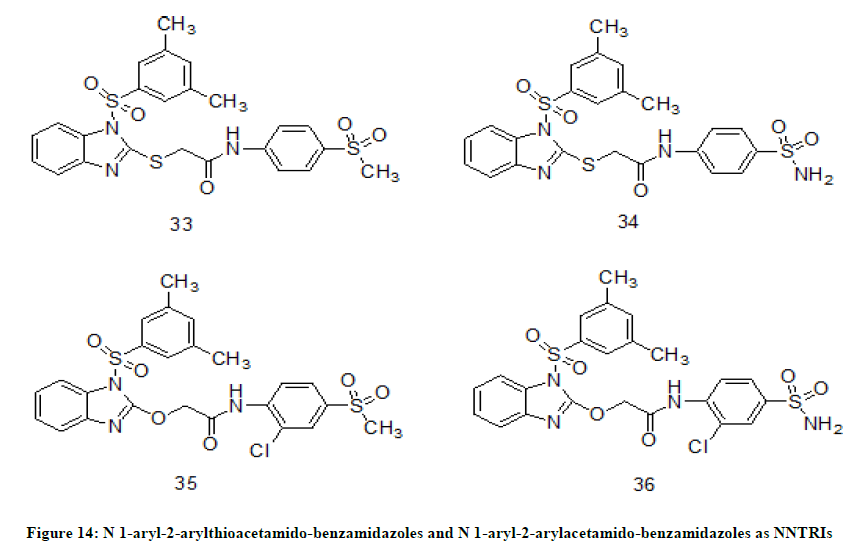
Figure 14: N 1-aryl-2-arylthioacetamido-benzamidazoles and N 1-aryl-2-arylacetamido-benzamidazoles as NNTRIs
Dihydrofuro[3,4-d]pyrimidine derivatives
D. Kang et.al. [24] have identified and synthesized dihydrofuro[3,4-d]pyrimidine as novel diarylpyrimidine (DAPY) derivatives and the compounds were proven to potent against wide range of HIV-1 strains. The lead compounds 37 (EC50=2.20 ± 0.67 nM), 38 (EC50=2.21 ± 0.61 nM), 39 (EC50(IIIB)=4.3 ± 0.7 nM) and 40 (EC50(IIIB)=2.6 ± 1.0 nM) (Figure 15) have shown excellent inhibition activity.
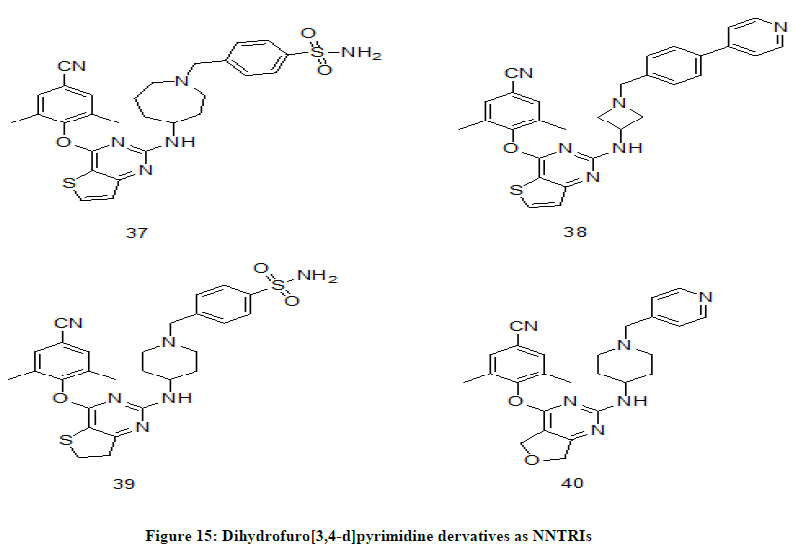
Figure 15: Dihydrofuro[3,4-d]pyrimidine dervatives as NNTRIs
1,4-Disubstituted 1,2,3-triazoles derivatives of diarylnicotinamide
1,4-Disubstituted 1,2,3-triazoles tailing was appended to diarylnicotinamide to afford series of novel HIV-1 non-nucleoside reverse transcriptase enzyme inhibitors [25]. The target NNTRIs were proven to be potent (EC50 0.02-1.77 μM) against wild-type and E138K mutant virus. Among the final compounds, compound 41, EC50(IIIB)=0.020 μM, EC50(E138K)=0.027 μM, CC50=180.90 μM) (42, EC50(IIIB)=0.020 μM, EC50(E138K)=0.014 μM, CC50=58.09 μM) and 43, (EC50(IIIB)=0.020 μM, EC50(E138K)=0.015 μM, CC50=40.15 μM) (Figure 16), are the most promising derivatives which have exhibited comparable potency with reference drug Etravirine against E138K mutant strain but with much lower cytotoxicity.
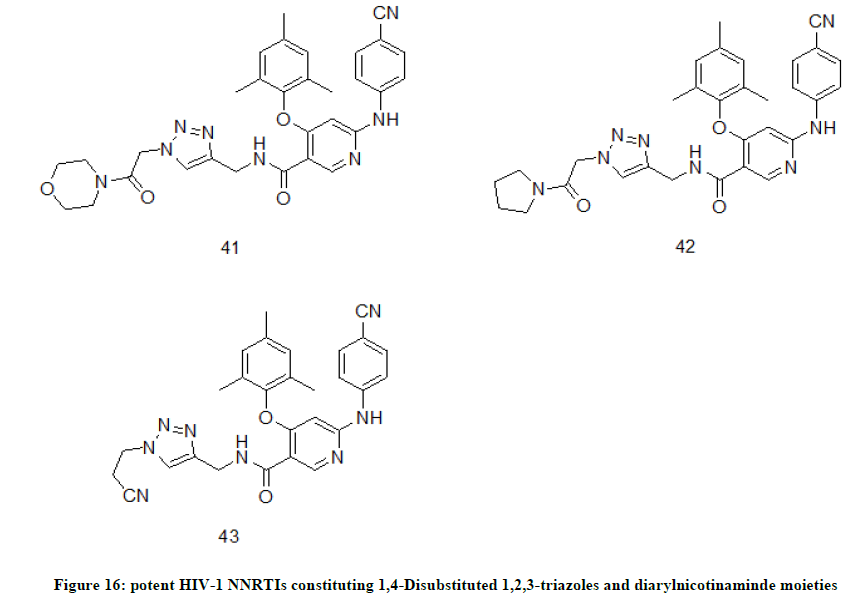
Figure 16: potent HIV-1 NNRTIs constituting 1,4-Disubstituted 1,2,3-triazoles and diarylnicotinaminde moieties
5-Hydroxypyrido[2,3-b]pyrazin-6(5H)-one derivatives
Recently dual inhibitor strategy has become an effective strategy for synthesis of anti-HIV drugs. In this context, 5-Hydroxypyrido[2,3-b]pyrazin-6(5H)-one derivatives were designed as a series of novel inhibiting agents for HIV-1 reverse transcriptase as well as integrase enzymes [26]. In the series of dual inhibitors, compounds 44 (IC50=1.77 μM) and 45 (IC501.85 μM) (Figure 17) have exhibited striking HIV-1 RT enzyme inhibition potency. While the compounds 46 (IC50=0.25) and 47 (IC50=0.22 μM) have exhibited promising inhibition activity against integrase. The pharmacology activity of the lead compounds was also supported by molecular docking studies of corresponding compounds.
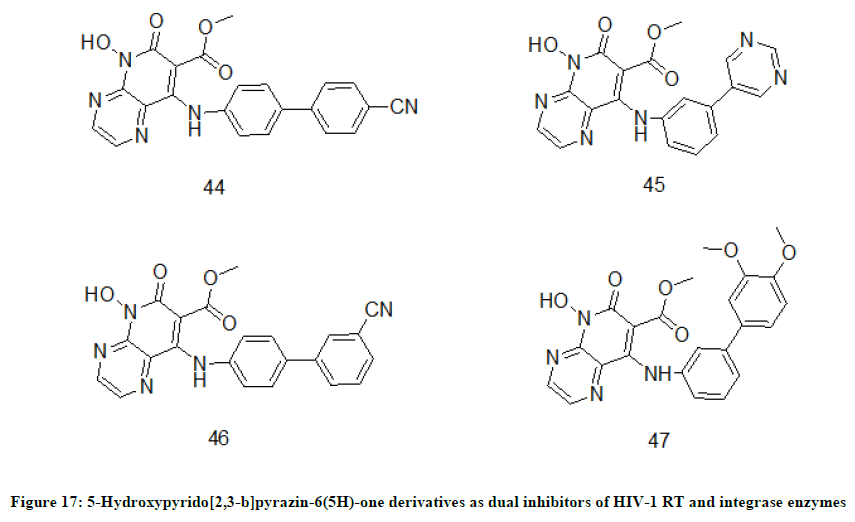
Figure 17: 5-Hydroxypyrido[2,3-b]pyrazin-6(5H)-one derivatives as dual inhibitors of HIV-1 RT and integrase enzymes
Magnesium chelated 2-Hydroxyisoquinoline-1,3(2H,4H)-diones
M. Billamboj et. al. [27] have planned and synthesized the magnesium chelated 2-Hydroxyisoquinoline-1,3(2H,4H)-diones. The chelates were evaluated for HIV ribonuclease H and integrase enzyme inhibition activity. In the corresponding antiviral activity compounds 48 and 49 (Figure 18) have shown efficient binding with magnesium cation in a 1: 1 stoichiometry.
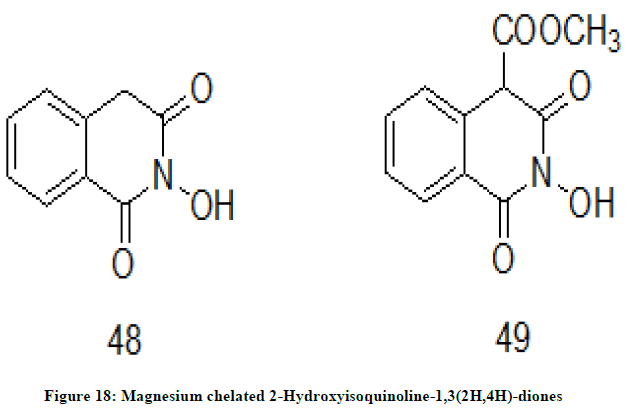
Figure 18: Magnesium chelated 2-Hydroxyisoquinoline-1,3(2H,4H)-diones
5,6,7,8-Tetrahydro-1,6-naphthyridine Derivatives
5,6,7,8-Tetrahydro-1,6-naphthyridine Derivatives were designed and prepared by K. Peese et. al. [28] targeting p75 (LEDGF/p75) binding site on HIV-1 integrase and LEDGF/p75 binding site of the integrase enzyme. In the prepared derivatives, compounds 50 (EC50=0.004 μM), 51(EC50 0.012 μM) and 52 (EC50=0.019 μM) (Figure 19) have shown best antiviral activity.
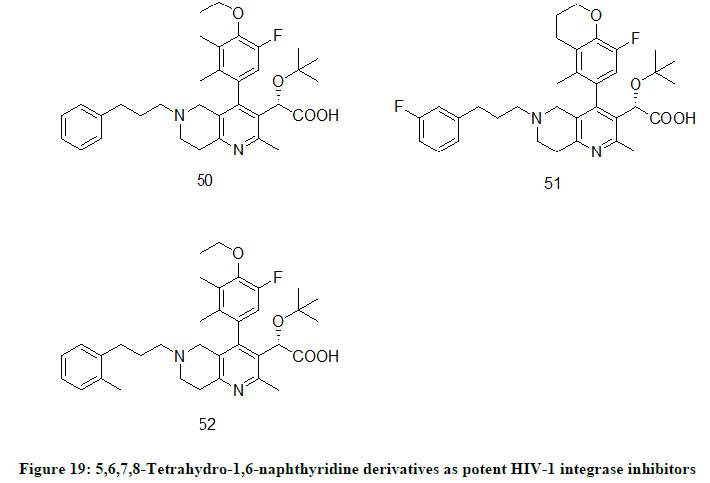
Figure 19: 5,6,7,8-Tetrahydro-1,6-naphthyridine derivatives as potent HIV-1 integrase inhibitors
Tetrazole derivatives of 7-(2H-Tetrazol-5-yl)-1H-indoles
A novel series of tetrazole derivatives, 7-(2H-Tetrazol-5-yl)-1H-indoles were synthesized and evaluated for antiviral activity [29]. Compounds 53 (EC50=34 nM), 54 (EC50=60 nM), 55 (EC50=120 nM) and 56 (EC50=0 nM) (Figure 20) were found to have best antiviral activity. Compound 56 represented as an example of N-methyl tetrazole that can acts as a prodrug for a free NH tetrazole-containing compound.
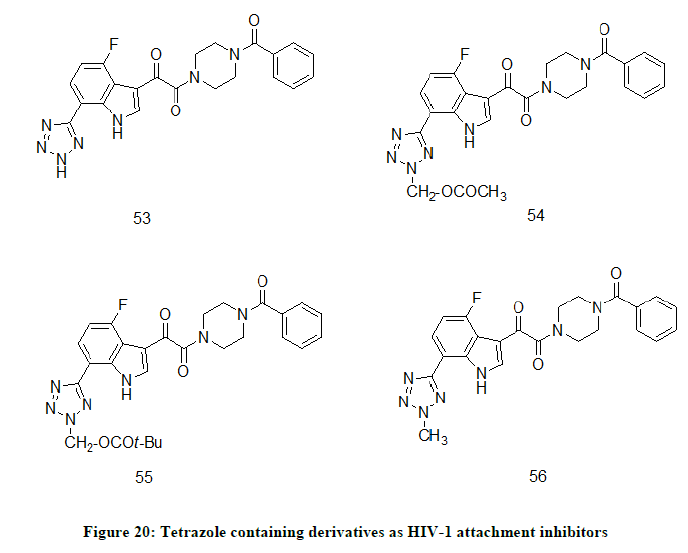
Figure 20: Tetrazole containing derivatives as HIV-1 attachment inhibitors
Sulfated polyanions
Sulfated polyanions are important scaffolds in anti-HIV activity and are good inhibitors of HIV-1 RNase H and reverse transcriptase enzymes [30]. Dextran, dextran sulfate, xylan polysulfate, DEAE-dextran, chondroitin sulfate, heparin etc. can be quoted as examples.
Illimaquinone derivatives
Illimaquinone 57, secondary metabolite (Figure 21) extracted from red sea sponges Smenospongia sp, synthetic derivatives of Illimaquinone, 6'-acetate illimaquinone 58 and 6'-methyl illimaquinone 59, Avarone A 60, Avarone B 61, Avarone E 62, Avarone F 63 are some of naturally occurring HIV-1 RNase inhibitors [31].
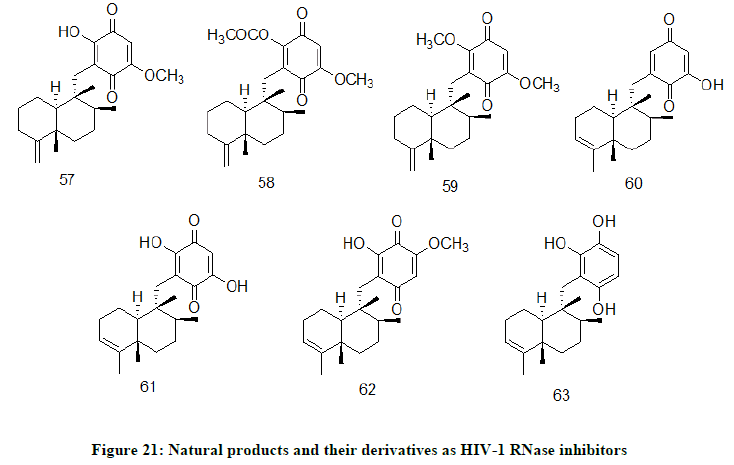
Figure 21: Natural products and their derivatives as HIV-1 RNase inhibitors
β-Thujaplicinol
β-thujaplicinol 64, (Figure 22) a natural compound derived from heartwood of cupressaceous trees has exhibited HIV-1 RNase H inhibition activity without affecting the DNA polymerase function of the enzyme [32].
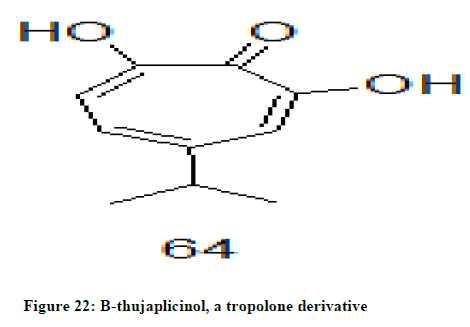
Figure 22: B-thujaplicinol, a tropolone derivative
Suramin
A well know antitrypanosomal agent, suramin 65 (Figure 23) was found to exhibit an excellent inhibitory activity on the enzyme reverse transcriptase using ethidium bromide [33].
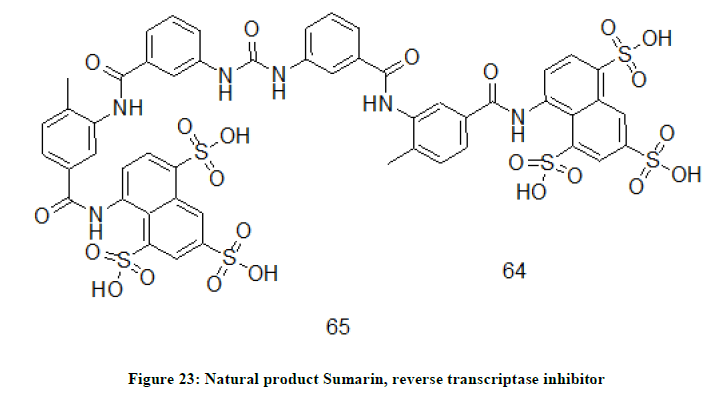
Figure 23: Natural product Sumarin, reverse transcriptase inhibitor
4′- and 2- modified 2′- Deoxyadenosine derivatives
4′Ed2FA (2′-deoxy-4′-C-ethynyl-2-fluoroadenosine) 66, a nucleoside derivative modified at 4′- and 2-positions of physiologic 2′- deoxyadenosine has been reported to have potent anti-HIV 1 properties including multidrug-resistant viruses and M184V HIV-1 mutant variants [34,35]. Alongside, its toxicity is reported to be lower than 4′EdA (2′-deoxy-4′-C-ethynyladenosine) (Figure 24). 4′Ed2ClA 67, chloro analog of compound 66 is active against all HIV-1s. However, activity of compound 67 is comparatively lower than that of 4′Ed2FA. Comparative study also indicated that the 3′-OH played important roles in the inhibiting activity against drug-resistant HIV-1s.
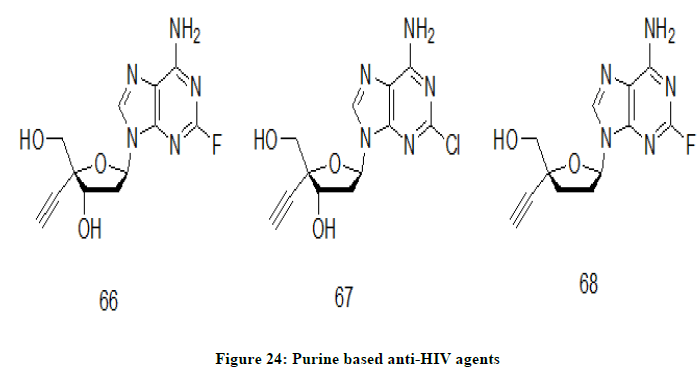
Figure 24: Purine based anti-HIV agents
FDA approved and Investigational HIV drugs
Investigational drugs are drugs under clinical trial in the different phases and some of them are utilized in successful treatment of HIV infection. These drugs are approved by U.S. Food and Drug Administration (FDA) after studying their effectiveness and adversity. According to the mode of action of investigational drugs, they have been classified into different classes.
Antifibrotics
The chemical drugs that reduce tissue scarring in the body are considered as anitfibrotics. Fibrosis is a pathological state in which excess deposition of fibrous tissue is seen. Losartan 69 and Telmisartan 70, (Figure 25) biphenyls derivatives are antifibrotic drugs used to treat high blood pressure and kidney damage caused by diabetes. HIV infection causes fibrosis in lymphoid tissues and there by damaging the immune system of the body [36-39]. Losartan is a tetrazole derivative of biphenyl whereas telminsartan is benzimidazole derivative of biphenyl moiety. Losartan improves immune system by reducing fibrosis. It may also aid in the reduction of latent HIV reservoirs [40-42].
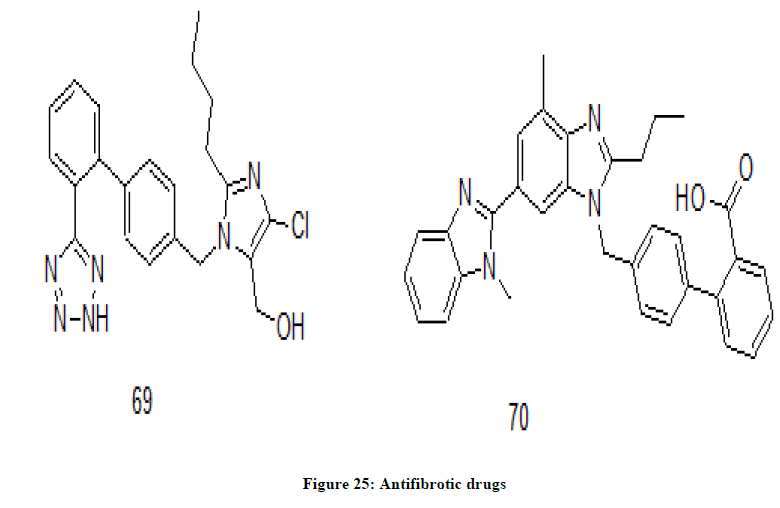
Figure 25: Antifibrotic drugs
CCR5 antagonists
Maravirok 71 and P140 72 belong to phenylpropylamine derivatives and are classified as CCR5 antagonists (Figure 26). These drugs attaches to CCR5 co-receptor protein on the surface of immune cells and prevents certain strains of HIV such as R5-tropic virus from attaching, enter or infect the cell [43,44] and thus prevents HIV from multiplying there by reduces the amount of HIV in the body. Because of quick absorbance properties of Maravirok in the body and accumulation of high concentrations of drug in tissues of vagina, cervix and rectum when taken as oral form, it is a possible medicine for HIV pre-exposure prophylaxis (PrEP).
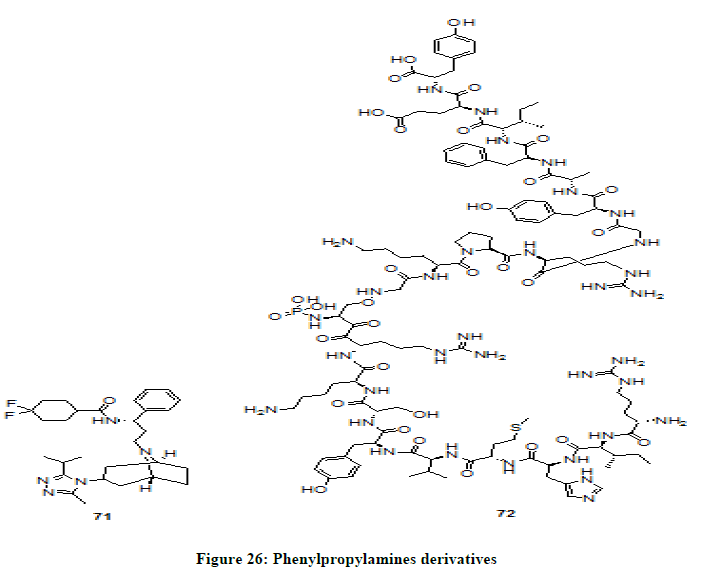
Figure 26: Phenylpropylamines derivatives
CD4 Attachment inhibitors
UB-421 and Fostemsavir 73 are classified as CD4 Attachment inhibitors. This class of drugs functions similar to that of CCR5 antagonists but the difference lies with respect to the cells that the drug attaches to. UB-421 attaches to a protein, CD4 receptor present on the surface of the immune cells. These drugs also prevent HIV particles to attach, enter or infect the cells in the body [45,46]. Fostemsavir (Figure 27), cyclohexanecarbodiimide and triazole derivative works by blocking the gp120 receptor of virus. The drug prevents initial viral attachment to CD4+ T cell and entry into the human cell and its method of action is a first for HIV drugs [47].
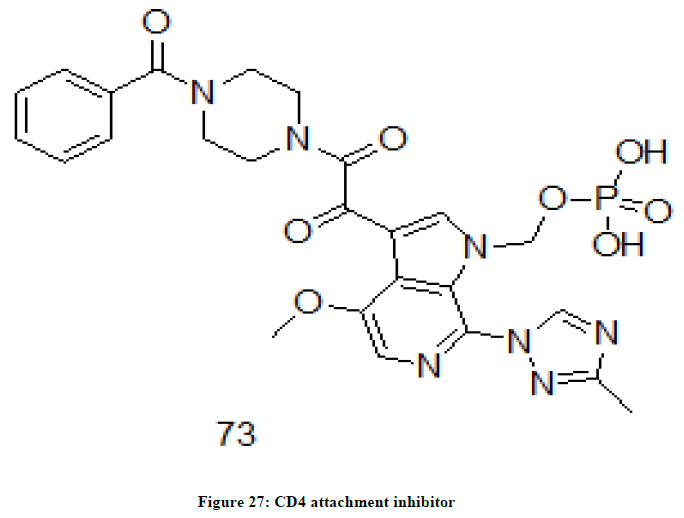
Figure 27: CD4 attachment inhibitor
CXCR4 antagonist
Plerixafor 74 and BL 8040 75 drugs are considered as CXCR4 antagonists (Figure 28) as they block the CXCR4 receptor and prevent its activation is a substance which blocks the CXCR4 receptor and prevent its activation. CXCR4 antagonists help to prevent metathesis by hindering the CXCR4 receptor activation. CXCR4 receptors are targeted by the antagonist since these are identified as co-receptor in HIV [48-50].
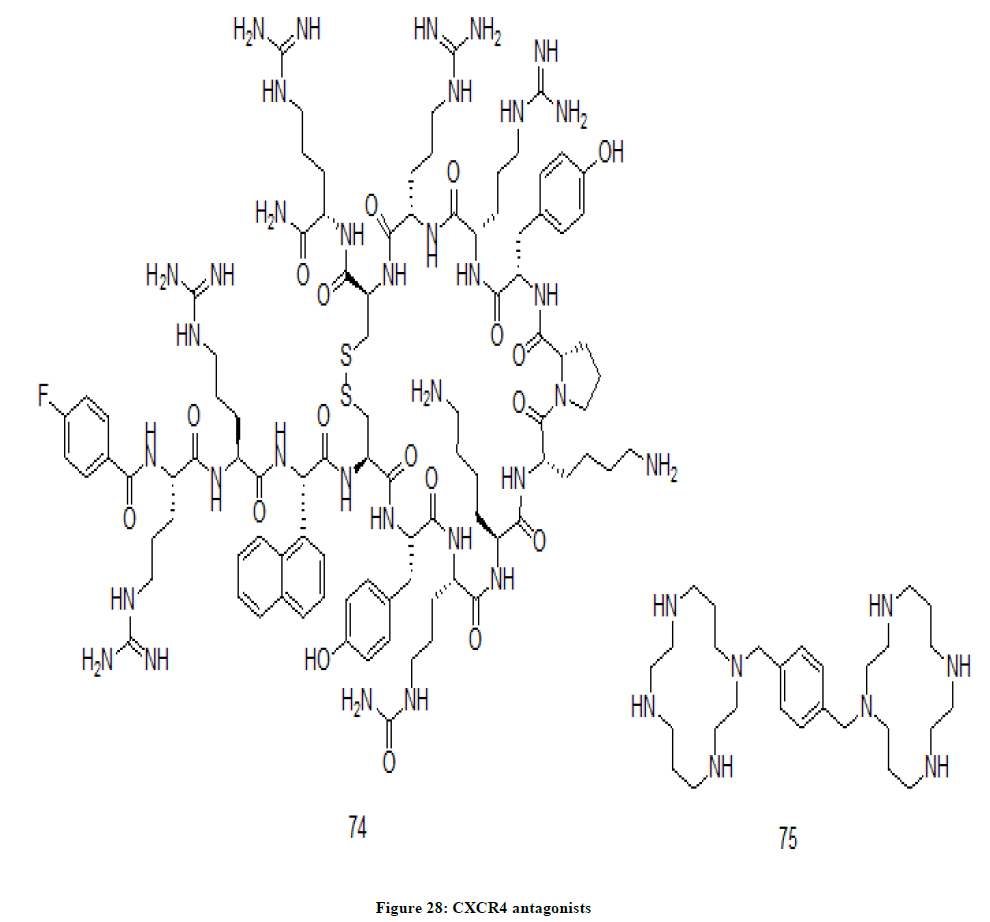
Figure 28: CXCR4 antagonists
Fusion inhibitors
A class of drugs called fusion inhibitors which fuse to the protein moieties on the outer surface of the HIV and inhibit the HIV particles from entering and infecting the immune cells. Albuvitride [51] and Enfuvitride belong to fusion inhibitors. Both the drugs are polypeptide derivatives. Albuvitride 76 (Figure 29) works by attaching to gp41, an envelope protein and there by blocking the HIV entry into the cells and infecting immune cells [52,53]. Enfuvitride functions in a similar fashion.
Figure 29: Fusion inhibitors
Immune modulators
Aldesleukin, chloroquine 77, hydroxychloroquine 78, CYT 107, Peginterferon Alfa-2a, Peginterferon Alfa-2b, Pyridostigmine 79 and Somatropin are categorized as immunomodulators (Figure 30). These chemical drugs help to boost and restore normal immune function. Aldesleukin is a derivative of a naturally occurring protein, interleukin-2 in the body as the latter aids in activation of immune system to make more immune cells and stimulates existing immune cells to fight opportunistic diseases [54-56].
Figure 30: Immunomodulators
Choroquine and hydroxychloroquine drugs are derivatives of 4-aminochloroquine moiety. These drugs can reduce hyperactivity of immune system and help to decrease HIV reservoirs and increase CD4 counts [57,58]. Peginterferon Alfa-2a and Peginterferon Alfa-2b are polypeptide drugs. Interferon alfa is a prepared naturally in the body that fights viral infections. Hence of a version is prepared known as peginterferon alfa-2a [59,60]. Peginterferon alfa-2a is administered along with other drugs to eliminate latent HIV which cannot be possible by ART alone [61,62]. Peginterferon Alfa-2b, Pyridostigmine and Somatropin have more or less similar functions in the body.
Integrase inhibitors
These chemical drugs block HIV enzyme integrase, and thereby preventing HIV from multiplying subsequently reducing viral load in the human body. These drugs are also called integrase inhibitors [63,64]. Some of the integrase inhibitors (Figure 31) which have been used for HIV treatment are under clinical trial such as bictegravir 80, cabotegravir 81, raltegravir 82, dalutegravir 83. Above quoted drugs are all fluorobenzene derivatives.
Figure 31: Chemical drugs as integrase inhibitors
Latency reversing agents
ART is ineffective in HIV treatment as the HIV particles go into the latent stage. In such case latent reversing agents are particularly beneficial. These chemical drugs are able to revoke the HIV virus present in immune cells (CD4) in latent stage. The activated HIV particles are neutralized by immune system or ongoing antiretroviral therapy (ART) [65].
Lefitolimod is categorized as a toll-like receptoragonist. Other latent reversing agents [66-68] (Figure 32) are panobinostat 84, vorinostat 85, valproic acid 86, tucidinostat 87 and romidepsin 88.
Figure 32: Latent reversing agents as HIV drugs
Maturation inhibitors
A category of chemical drugs which inhibit the maturation of the virus are known as maturation drugs such as bevirimat 89 (Figure 33). These drugs inhibit the final step in the processing of the HIV-1 gag protein by binding to it and results in the formation of noninfectious, immature virus particles and such viruses are incapable of infecting other cells [69-71].
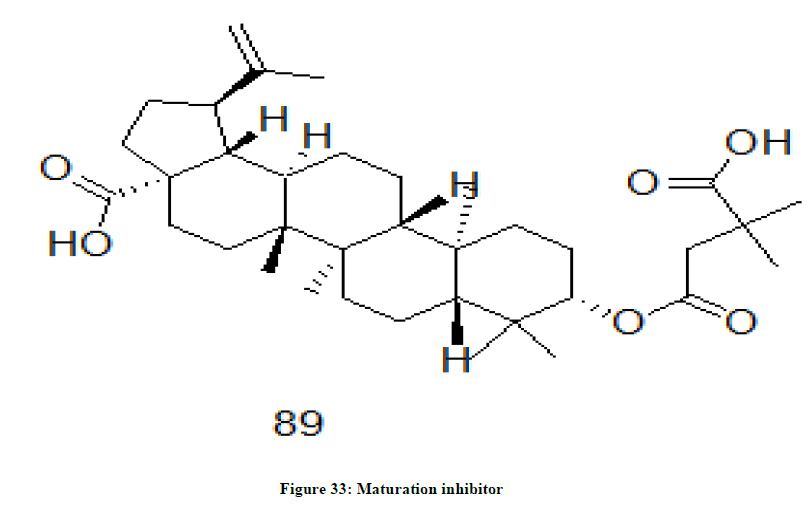
Figure 33: Maturation inhibitor
Microbicides
Dapivirine 91 and tenofovir 90 are microbicides (Figure 34). These chemical drugs prevent sexual transmission of HIV and used as a topical microbicide. These products are designed so as to prevent HIV during sexual activities where these topical microbicides are applied to the body parts that are engaged in sexual activities [72-74]. Dapivirine is a pyrimidine derivative whereas tenofovir is a nucleotide derivative.
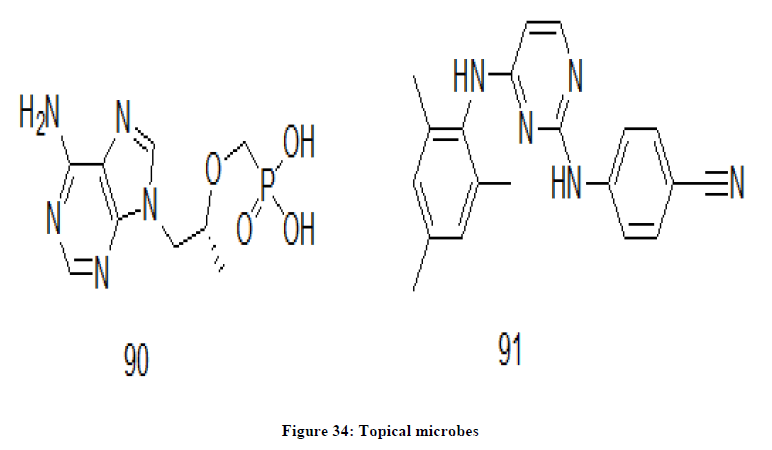
Figure 34: Topical microbes
Non-nucleoside reverse transcriptase inhibitors (NNRTIs)
NNRTIs are HIV-1 specific and inactive against other retroviruses. Reverse transcriptase enzyme is must for the HIV particles to multiply in the human body. NNRTIs are the chemical drugs that attach to reverse transcriptase and thereby blocking it and subsequently reducing the amount of HIV in the body [75,76]. In this category of drugs, elsulfavirine 92, doravirine 93, rilpivirine 94, efavirenz 95, etravirine and nevirapine can be quoted as some of the examples (Figure 35).
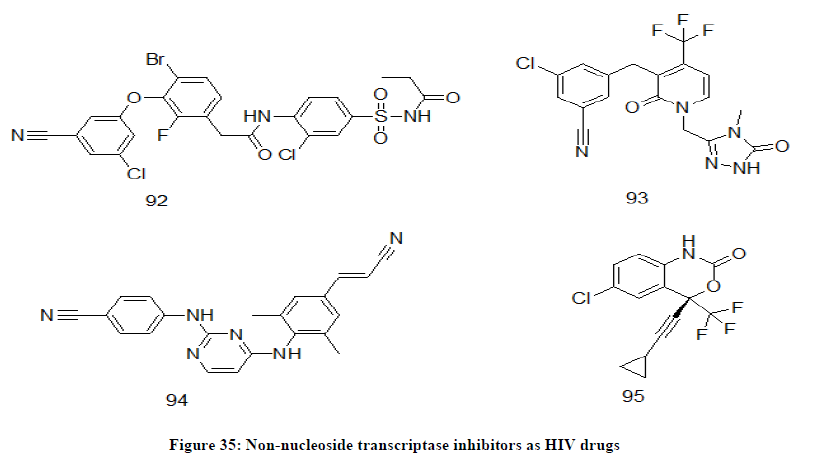
Figure 35: Non-nucleoside transcriptase inhibitors as HIV drugs
Nucleoside reverse transcriptase inhibitors (NRTIs)
NRTIs are chemical drugs of nucleoside analogues. These drugs have a nucleoside sub unit in them. They block an essential enzyme, reverse transcriptase of HIV for its replication in the human body. Blockage of the particular inhibits multiplication of the HIV and thus its viral load reduces in the body. A few NRTIs may be named as Abacavir 96, didanosine 97, emtricitabine 98 (Figure 36), emtriva, epivir, lamivudine, Retrovir, videx, viread, ziagen, zidovudine etc. Abacavir is a nucleoside reverse transcriptase inhibitor which has guanosine unit. It inhibits HIV reverse transcriptase and thus blocking viral replication [77,78].
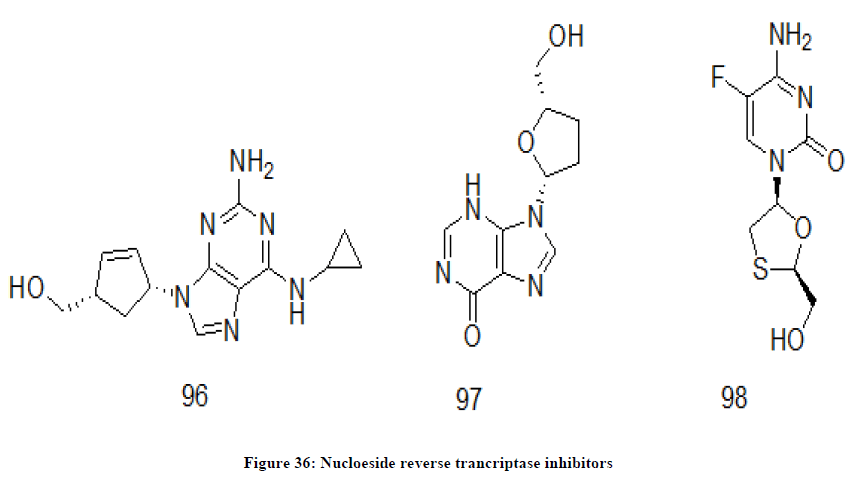
Figure 36: Nucloeside reverse trancriptase inhibitors
Opportunistic infections and co-infections
These are the infections which occur more frequently when the immune system is weakened such as in 3rd stage of HIV infection [79]. These are generally noninfectious if the immune system is strong enough. HSV and VSV can be quoted as examples of opportunistic infections of HIV. Some of the chemical drugs (Figure 37) which have been used in the treatment of opportunistic infections are Butoconazole Nitrate 99, femciclovir 100, Acyclovir 101, daclatasvir 102, ribavirin 103, ganciclovir, sofosbuvir etc.
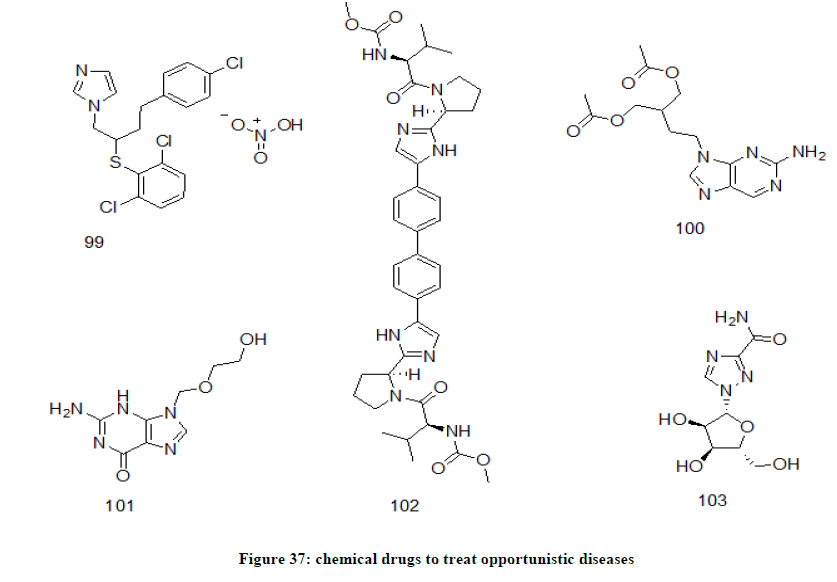
Figure 37: chemical drugs to treat opportunistic diseases
Acyclovir is nucleoside derivative of glycol. It is an antiviral used to treat herpes simplex virus (HSV) infections and varicella virus diseases (VZV). Butoconazole, imidazole derivative is used to treat opportunistic infections like vulvovaginal candidiasis. Daclatasvir is biphenyl and imidazole derivative used to treat chronic hepatitis C virus infection. Femciclovir is diester derivative of purine and is useful in the treatment of two types of herpes simplex virus (HSV) and also for VZV infection. Ribavirin is a synthetic nucleoside derivative of ribofuranose and is used in the treatment of hepatitis C virus and similar viruses. Ribavirin inhibits viral RNA synthesis and thus inhibiting normal viral replication.
Pharmacokinetic enhancers
The chemical drugs which increase the concentration of other HIV medicines in the blood are known as pharmacokinetic enhancers. Such drugs help in the breakdown of the other HIV drugs in the body and enhance their effectiveness. Some of the pharmacokinetic enhancers (Figure 38) are Cobicistat 104 and Ritonavir 105. Cobicistat is a thiazole and carbamate derivative that inhibits CYTOCHROME P450 CYP3A enzyme. Cobicistat is always used in the combination with other HIV drugs as it is not an antiviral [80].
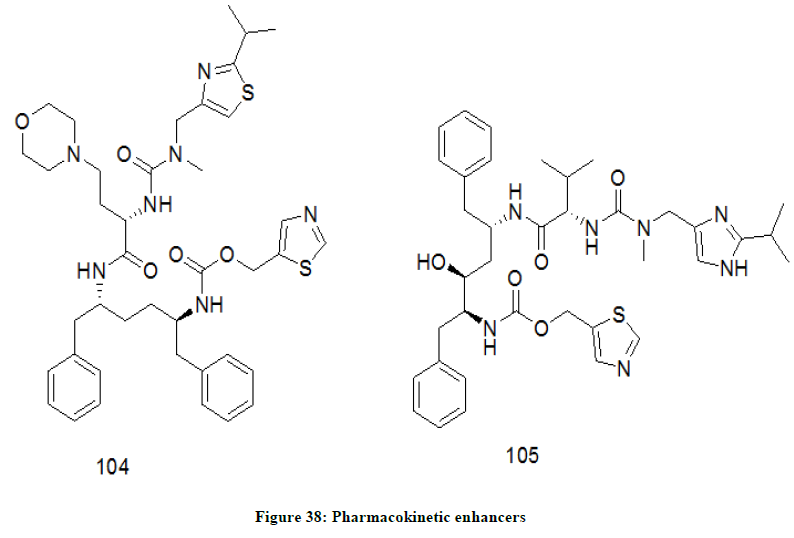
Figure 38: Pharmacokinetic enhancers
Post-attachment inhibitors
Post-attachment drugs inhibits the entry of HIV particle into the human cells by binding to the CCR5 and CXCR4 co-receptors after HIV binds to the CD4 receptor (primary receptor) [81] on the surface of a CD4 cell. The chemical drug Ibalizumab is categorized as post-attachment inhibitor and is a monoclonal antibody. Post-attachment inhibitors are one of the sub-classes of HIV entry inhibitors.
Protease inhibitors
These chemical drugs inhibit the protease enzyme of HIV and reduce the HIV replication in the human cells [82]. Some of the protease inhibitors (Figure 39) are atazanavir 106, Saquinavir 107, fosamprenavir 108, tipranavir etc.
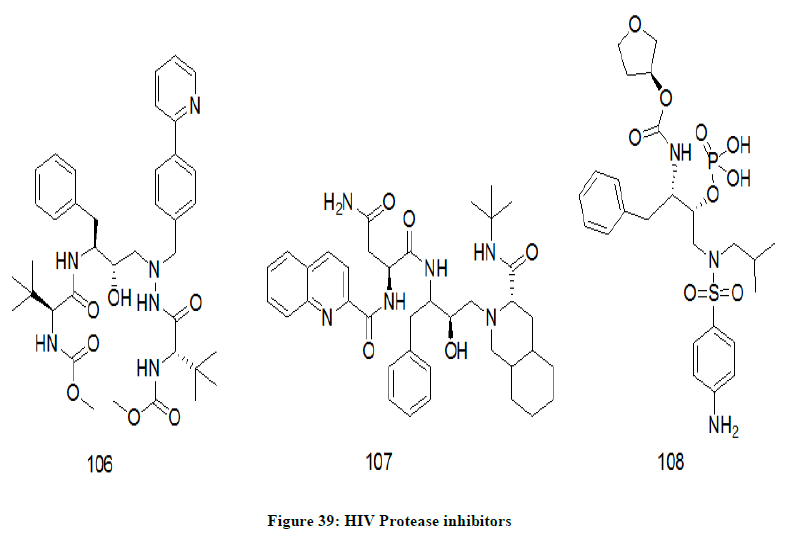
Figure 39: HIV Protease inhibitors
Rev inhibitors
Rev is a trans activating protein which is essential to the regulation of HIV-1 protein expression. The Rev gene present in the HIV is responsible for translation. In the translational process Rev gene is involved in export of unspliced and incompletely spliced mRNAs [83]. ABX464 109 (Figure 40) is an example of Rev Inhibitor and it is chloroquinoline derivative.
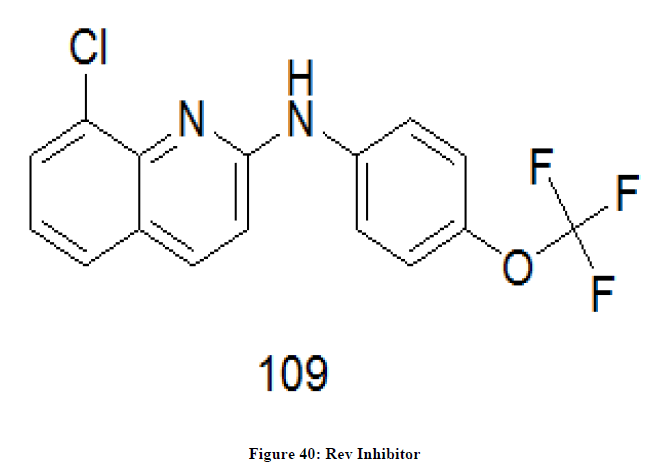
Figure 40: Rev Inhibitor
gp120 attachment inhibitor
The chemical drugs that bind to the gp120 protein of outer surface of HIV and inhibits the HIV from getting into and infecting immune cells [84,85]. Fostemsavir 14 (Figure 6) is an example of gp 120 attachment inhibitor [86,87]. Fostemsavir is a prodrug and gets converted into active form, temsavir 110 (Figure 41) in the body.
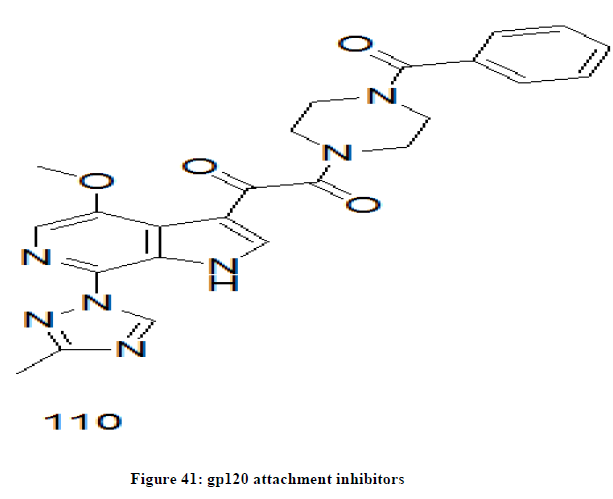
Figure 41: gp120 attachment inhibitors
Conclusion
The HIV virus is a type of retrovirus by its literal meaning seemingly simple as that of other viruses but difference lies in its menacing action on the immune cells of the human body as HIV weakens immune system by decreasing CD4-T cells. Reduction of CD4-T cells makes human immune system weaker and thereby allowing the opportunistic infections to collapse the immune system completely. The natural products, its derivatives and large classes of chemical drugs have been synthesized for the prevention, cure and control of HIV infection, and opportunistic infections. Mode of action of all these drugs is based on the inhibition of a particular type of HIV protein which would be essential for its replication, maturation, attachment to the immune cells, or entry into the cell etc. Some of the drugs work based upon the stages of the life cycle of HIV virus. After all the infection, a few class of chemical drugs synthesized to prevent the menace of opportunistic infections thereby increasing the life span of the HIV infected person.
The HIV virus is a type of retrovirus by its literal meaning seemingly simple as that of other viruses but difference lies in its menacing action on the immune cells of the human body as HIV weakens immune system by decreasing CD4-T cells. Reduction of CD4-T cells makes human immune system weaker and thereby allowing the opportunistic infections to collapse the immune system completely. The natural products, its derivatives and large classes of chemical drugs have been synthesized for the prevention, cure and control of HIV infection, and opportunistic infections. Mode of action of all these drugs is based on the inhibition of a particular type of HIV protein which would be essential for its replication, maturation, attachment to the immune cells, or entry into the cell etc. Some of the drugs work based upon the stages of the life cycle of HIV virus. After all the infection, a few class of chemical drugs synthesized to prevent the menace of opportunistic infections thereby increasing the life span of the HIV infected person.
The HIV virus is a type of retrovirus by its literal meaning seemingly simple as that of other viruses but difference lies in its menacing action on the immune cells of the human body as HIV weakens immune system by decreasing CD4-T cells. Reduction of CD4-T cells makes human immune system weaker and thereby allowing the opportunistic infections to collapse the immune system completely. The natural products, its derivatives and large classes of chemical drugs have been synthesized for the prevention, cure and control of HIV infection, and opportunistic infections. Mode of action of all these drugs is based on the inhibition of a particular type of HIV protein which would be essential for its replication, maturation, attachment to the immune cells, or entry into the cell etc. Some of the drugs work based upon the stages of the life cycle of HIV virus. After all the infection, a few class of chemical drugs synthesized to prevent the menace of opportunistic infections thereby increasing the life span of the HIV infected person.
Acknowledgment
I extend sincere thanks for SCI-HUB for providing opportunity to access a large number of full length scientific research papers. I acknowledge heartfelt thanks to my better half A. Premalatha.
References
- E. De Clercq, A. Holy, Nat. Rev. Drug Discovery, 2005, 4, 928.
- J. Balzarini, A. Holy, J. Jindrich, L. Naesens, R. Snoeck, D. Schols, E. De Clercq, Antimicrob. Agents Chemother., 1993, 37, 332.
- R. Heijtink, J. Kruining, G. De Wilde, J. Balzarini, E. De Clercq, S. Schalm, Antimicrob. Agents Chemother., 1994, 38, 2180.
- M. Luo, E. Groaz, S. De Jonghe, D. Schols, P. Herdewijn, Chem. Biodiversity, 2018.
- A. Regueiro-Ren, J. J. Swidorski, Z. Liu, Y. Chen, N. Sin, S. Y. Sit, J. Chen, B. L. Venables, J. Zhu, B. Nowicka-Sans, T. Protack, Z. Lin, B. Terry, H. Samanta, S. Zhang, Z. Li, J. Easter, B.R. Beno, V. Arora, X.S. Huang, S. Rahematpura, D.D. Parker, R. Haskell, K.S. Santone, M.I. Cockett, M. Krystal, N.A. Meanwell, S. Jenkins, U. Hanumegowda, I.B. Dicker, J. Med. Chem., 2018, 61, 7289
- H. Sinokrot, T. Smerat, A. Najjar, R. Karaman, Molecules, 2017, 22, 1736.
- Y. Hamada, Bioorg. Med. Chem. Lett., 2017, 27, 1627.
- B. Huang, X. Liu, Y. Tian, D. Kang, Z. Zhou, D. Daelemans, E. De Clercq, C. Pannecouque, P. Zhan, X. Liu, Bioorg. Med. Chem. Lett., 2018, 28, 1348.
- M. Hurevich, A. Swed, S. Joubran, S. Cohen, N.S. Freeman, E. Britan-Rosich, L. Briant-Longuet, M. Bardy, C. Devaux, M. Kotler, A. Hoffman, C. Gilon, Bioorg. Med. Chem.,2010, 18, 5754.
- T. Wang, Y. Ueda, Z. Zhang, Z. Yin, J. Matiskella, B.C. Pearce, Z. Yang, M. Zheng, D.D. Parker, G.A. Yamanaka, Y.F. Gong, H.T. Ho, R.J. Colonno, D.R. Langley, P.F. Lin, N.A. Meanwell, J.F. Kadow, J. Med. Chem., 2018, 61, 308.
- B. Huang, D. Kang, J. Yang, P. Zhan, X. Liu, a patent evaluation of US20140378443A1. Expert Opin. Ther. Pat., 2016, 26, 281.
- Z. Wang, D. Kang, M. Chen, G. Wu, D. Feng, T. Zhao, Z. Zhou, Z. Huo, L. Jing, X. Zuo, D. Daelemans, E. D. Clercq, C. Pannecouque, P. Zhan, X. Liu, Chem. Bio. Drug Design, 2018.
- A.K. Ghosh, R.D. Jadhav, H. Simpson, S. Kovela, H. Osswald, J. Agniswamy, Y.F. Wang, S.I. Hattori, I.T. Weber, H. Mitsuya, Eur. J. Med. Chem., 2018, 10.1016/j.ejmech.2018.09.046
- A.K. Ghosh, P.R. Nyalapatla, S. Kovela, K.V. Rao, M. Brindisi, H.L. Osswald, M. Amano, M. Aoki, J. Agniswamy, Y.F. Wang, I.T. Weber, H. Mitsuya, J. Med. Chem., 2018, 61, 4561.
- A.K. Ghosh, B.D. Chapsal, I.T. Weber, H. Mitsuya, Acc. Chem. Res., 2008, 41, 78.
- A.K. Ghosh, P.R. Sridhar, N. Kumaragurubaran, Y. Koh, I.T. Weber, H. Mitsuya, ChemMedChem., 2006, 1, 939.
- X. Bai, Z. Yang, M. Zhu, B. Dong, L. Zhou, G. Zhang, J. Wang, Y. Wang, Eur. J. Med. Chemi., 2017, 137, 30.
- J.S.G. Montaner, V.D. Lima, R. Barrios, B. Yip, E. Wood, T. Kerr, K. Shannon, P.R. Harrigan, R.S. Hogg, P. Daly, P. Kendall, ancet, 2010, 376, 532.
- N. Lohse, A.B. Hansen, J. Gerstoft, N. Obel, J. Antimicrob. Chemother., 2007, 60, 461.
- A. Wlodawer, J. Vondrasek, Annu. Rev. Biophys. Biomol. Struct., 1998, 27, 249.
- A.K. Ghosh, J.N. Williams, R.Y. Ho, H.M. Simpson, S.I. Hattori, H. Hayashi, J. Agniswamy, Y.F. Wang, I.T. Weber, H. Itsuya, J. Med. Chem.,2018.
- T. Frączek, R. Kamiński, A. Krakowiak, E. Naessens, B. Verhasselt, P. Paneth, J. Enzyme Inhib. Med. Chem., 2018, 33, 9.
- A.M. Monforte, L.D. Luca, M.R. Buemi, F.E. Agharbaoui, C. Pannecouque, S. Ferro, Bioorg. Med. Chem., 2018, 26, 661.
- D. Kang, H. Zhang, Z. Wang, T. Zhao, T. Ginex, Francisco, J. Luque, Y. Yang, G. Wu, D. Feng, F. Wei, J. Zhang, E.D. Clercq, C. Pannecouque, C.H. Chen, K.H. Lee, N.A. Murugan, T.A. Steitz, P. Zhan, X. Liua, J. Med. Chem., 2019.
- Y. Tian, Z. Liu, J. Liu, B. Huang, D. Kang, H. Zhang, E. De Clercq, D. Daelemans, C. Pannecouque, K.H. Lee, C.H. Chen, P. Zhan, X. Liu, Eur. J. Med. Chem., 2018, 151, 339.
- L. Sun, P. Gao, G. Dong, X. Zhang, X. Cheng, X. Ding, X. Wang, D. Daelemans, E. De Clercq, C. Pannecouque, L.M. Arias, P. Zhan, X. Liu, Eur. J. Med. Chem., 2018, 155, 714.
- M. Billamboz, F. Bailly, C. Lion, N. Touati, H. Vezin, C. Calmels, M.L. Andreola, F. Christ, Z. Debyser, P. Cotelle, J. Med. Chem.,2011, 54, 1812.
- K. Peese, C. Allard, T. Connolly, B. Johnson, C. Li, M. Patel, M. Sorensen, M. Walker, N.A. Meanwell, B. McAuliffe, B. Minassian, M. Krystal, D. Parker, H.A. Lewis, K. Kish, P. Zhang, R.T. Nolte, J. Simmermacher, S. Jenkins, C. Cianci, B.N. Naidu, J. Med. Chem., 2019.
- K.S. Yeung, Z. Qiu, Z. Yang, L. Zadjura, C. J. D’Arienzo, M.R. Browning, S. Hansel, X.S. Huang, B.J. Eggers, K. Riccardi, P.F. Lin, N.A. Meanwell, J.F. Kadow, Bioorg. Med. Chem. Lett., 2013, 23, 209.
- K. Moelling, T. Schulze, H. Diringer, J. Virol., 1989, 63, 5489.
- M.M.A. Mitry, N.S. Abdou, R.A.T. Serya, Arch. Pharmaceut. Sci., 2018, 2, 22.
- E. De Clercq, Cancer Lett., 1979, 8, 9.
- H. Ohrui, Chem. Rec., 2006, 6,133.
- H. Ohrui, S. Kohgo, H. Hayakawa, E. Kodama, M. Matsuoka, T. Nakata, H. Mitsuya, Nucleos. Nucleot. Nucl.,2007, 26, 1543.
- M. Zeng, P.J. Southern, C.S. Reilly, G.J. Beilman, J.G. Chipman, T.W. Schacker, A.T. Haase, PLoS Pathog., 2012, 8, e1002437.
- M. Zeng, A.J. Smith, S.W. Wietgrefe, P.J. Southern, T.W. Schacker, C.S. Reilly, J.D. Estes, G.F. Burton, G. Silvestri, J.D. Lifson, J.V. Carlis, A.T. Haase, J. Clin. Invest., 2011, 121, 998.
- D. C. Douek, Top Antivir. Med., 2013, 21,128.
- H. Hatano, Curr. Opin. HIV AIDS, 2013, 8, 211.
- A.P. Andonopoulos, G. Tzanakakis, Rheumatol Int., 1993, 12, 255-7.
- https://www.niaid.nih.gov/clinical-trials/life-hiv-study.
- E. Bruzzesi, I. Sereti, Curr. Top. Microbiol. Immunol., 2018.
- W. Wienen, M. Entzeroth, J.C.A. Van Meel, J. Stangier, U. Busch, T. Ebner, J. Schmid, H. Lehmann, K. Matzek, J. Kempthorne-Rawson, V. Gladigau, N.H. Haue, Cardiovascular Drug Reviews, 2006, 18, 127.
- O. A. Milan, M. Sylla, M. E. Far, G. B. Jasmin, A. Haidara, J. Blackburn, A. Chamberland, C. L. Tremblay, Antimicrob. Agents Chemother., 2014, 58, 7565.
- Q. Tan, Y. Zhu, J. Li, Z. Chen, G.W. Han, I. Kufareva, T. Li, L. Ma, G. Fenalti, J. Li, W. Zhang, X. Xie, H. Yang, H. Jiang, V. Cherezov, H. Liu, R.C. Stevens, Q. Zhao, B. Wu, Science, 2013, 341, 1387.
- S. Ugolini, I. Mondor, P.W. Parren, D.R. Burton, S.A. Tilley, P.J. Klasse, Q.J. Sattentau, J. Exp. Med., 1997, 20, 1287.
- N.A. Meanwell, M.R. Krystal, B. Nowicka-Sans, D.R. Langley, D.A. Conlon, M.D. Eastgate, D.M. Grasela, P. Timmins, T. Wang, J.F. Kadow, J. Med. Chem., 2017, 61, 62.
- P. Cahn, V. Fink, P. Patterson, Curr. Opin. HIV AIDS, 2018, 13, 1.
- J.C. Knight, Med. Chem. Comm., 2012, 3, 1039.
- T. Murakami, S. Kumakura, T. Yamazaki, R. Tanaka, M. Hamatake, K. Okuma, W. Huang, J. Toma, J. Komano, M. Yanaka, Y. Tanaka, N. Yamamoto, Antimicrob. Agents Chemother., 2009, 53, 2940.
- M. Steinberg, M. Silva, Clin. Ther., 2010, 32, 821.
- R.M. Gulick, C. Flexner, Annu Rev Med., 2019, 70, 137.
- H. Chong, X. Yao, C. Zhang, L. Cai, S. Cui, Y. Wang, Y. He, PLoS ONE, 2012, 7, e32599,
- M. Boyd, S. Pett, Aust. Prescr. 2008, 31, 66.
- J. Naidoo, D.B. Page, J.D. Wolchok, Br. J.Cancer 2014, 111, 2214.
- V. Brinkmann, M.D. Davis, C.E. Heise, R. Albert, S. Cottens, R. Hof, C. Bruns, E. Prieschl, T. Baumruker, P. Hiestand, C.A. Foster, M. Zollinger, K.R. Lynch, J. Biol. Chem., 2002, 277, 21453.
- R. Paredes, J.L.B. de Quiros, F.E. -Cruz, B. Clotet, H. Lane, AIDS Rev. 2002, 4, 36.
- A. Savarino, I.L. Shytaj, Retrovirology, 2015, 12, 51.
- S.M. Murray, C.M. Down, D.R. Boulware, W.M. Stauffer, W.P. Cavert, T.W. Schacker, J.M. Brenchley, D.C. Douek, J. Virol., 2010, 84, 12082.
- S. Zeuzem, S.V. Feinman, J. Rasenack, E.J. Heathcote, M.Y. Lai, E. Gane, J. O'Grady, J. Reichen, M. Diago, A. Lin, J. Hoffman, M.J. Brunda, N. Engl. J. Med., 2000, 343, 1666.
- K. Gibbert, J.F. Schlaak, D. Yang, U. Dittmer, Br. J. Pharmacol., 2013, 168, 1048.
- R.T. Chung, J. Andersen, P. Volberding, G.K. Robbins, T. Liu, K.E. Sherman, M.G. Peters, M.J. Koziel, A.K. Bhan, B. Alston, D. Colquhoun, T. Nevin, N. Engl. J. Med., 2004, 351, 451.
- T. Piratvisuth, Hepatol. Int., 2008, 2, 140.
- E. Wong, N. Trustman, A. Yalong, J. Am. Acad. Pas, 2016, 29, 36.
- K. Shimura, E. N. Kodama, Antivir. Chem. Chemother., 2009, 20, 79.
- R.F. Siliciano, W.C. Greene, Cold Spring Harb. Perspect. Med.,2011, a007096.
- T.A. Rasmussen, M. Tolstrup, A. Winckelmann, L. Ostergaard, O.S. Sogaard Hum. Vaccin. Immunother., 2013, 9, 790.
- K. Bashiri, N. Rezaei, M. Nasi, A. Cossarizza, Immunol. Lett., 2018, 196, 135.
- A.M. Spivak, V. Planelles, Annu. Rev. Med., 2018, 69, 421.
- P.W. Keller, C.S. Adamson, J.B. Heymann, E.O. Freed, A.C. Steven, J. Virol., 2010, 85, 1420.
- K. Salzwedel, D.E. Martin, M. Sakalian, AIDS Rev., 2007, 9, 162.
- C.S. Adam, K. Salzwedel, E.O. Freed. Expert Opin. Ther. Targets, 2009, 13, 895.
- A.A. Waheed, E.O. Freed, AIDS Res. Hum. Retroviruses, 2012, 28, 54
- J. Obiero, P.G. Mwethera, C.S. Wiysonge, Cochrane Database Syst Rev., 2012, 13 CD007961.
- M.L. Cottrell, A.D.M. Kashuba, J. Clin. Pharmacol., 2014, 54, 603.
- R.J. Shattock, Z. Rosenberg, Cold Spring Harb. Perspect. Med., 2012, 2, a007385.
- S.M. Drake, J. Antimicrob. Chemother., 2000, 45, 4, 417.
- A. Sosnik, D.A. Chiappetta, A.M. Carcaboso, J. Controll. Release, 2009, 138, 2.
- T. Wu, J. Zhang, M. Geng, S.J. Tang, W. Zhang, J. Shu, Scientific Reports, 2017, 7, 1
- B.C. Zanoni, R.T. Gandhi, Infect. Dis. Clin. North America, 2014, 28, 501.
- L. Xu, M. C. Desai, Curr. Opin. Investig. Drugs, 2009, 10, 775.
- M.D. Swanson, H.C. Winter, I.J. Goldstein, D.M. Markovitz, J. Biol. Chem., 2010, 285, 8646.
- J.M. Jacobson, D.R. Kuritzkes, E. Godofsky, E. DeJesus, J.A. Larson, S.P. Weinheimer, S.T. Lewis, Antimicrob. Agents. Chemother., 2009, 53, 450.
- H. Adachi, Y. Takahashi, M. Kyo, T. Sato, Y. Nishimura, Lett. Drug Design Discovery, 2006, 3, 71.
- M.B. Feinberg, R.F. Jarrett, A. Aldovini, R.C. Gallo, W.F. Staal, Cell, 1986, 46, 807.
- J. Kadow, H. G. Wang, P. F. Lin, Curr. Opin. Investig. Drugs, 2006, 7, 721.
- R.E. Nettles, D. Schurmann, L. Zhu, J. Infect. Dis., 2012, 206, 1002.
- I. Mondor, S. Ugolini, Q.J. Sattentau, J. Virol., 1998, 72, 3623.



