Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 3
Ciprofloxacin: A Two Step Process
Veera Reddy Arava* and Pailla Umareddy
R & D Centre Suven Life Sciences Ltd, Plot No #18, Phase-III, Jeedimetla, Hyderabad-500055, India
- *Corresponding Author:
- Veera Reddy Arava
R & D Centre Suven Life Sciences Ltd
Plot No #18, Phase-III, Jeedimetla, Hyderabad-500055, India
Abstract
Evolution of different syntheses of ciprofloxacin are described. A high yielding two step synthesis from 2,4-dichloro-5-fluoro benzoylchloride is described.
References
Find Lawyer in Washington Blog | Places to Visit in Croatia Find Lawyer in Kentucky Blog | Places to Visit in Hungary Real Estate in South Africa Blog | Places to Visit in Indonesia Casino Sites in Russia Blog | Places to Visit in Iran Blog | Places to Visit in Italy Places to Visit in USA Blog | Places to Visit in Jordan Find Lawyer in Minnesota Blog | Places to Visit in Japan Mortgage info in Miami Blog | Places to Visit in South Korea Places to Visit in UK Blog | Places to Visit in Mexico Best Betting Sites in Cyprus Blog | Places to Visit in Malaysia Blog | Places to Visit in NetherlandsKeywords
2,4-Dichloro-5-fluoro benzoylchloride, 3-Dimethylamino-acrylic acid methyl ester, Piperazine, Cyclopropylamine, One pot
Introduction
Ciprofloxacin (1) is used to treat a wide variety of infections. Ciprofloxacin is broad based antibiotic of the fluoroquinoline class. It is active against both Gram-positive and Gram-negative bacteria (Figure 1). It functions by inhibiting DNA gyrase and a type-II and type-IV topoisomerases necessary to separate bacterial DNA, there by inhibiting cell division. The drug was invented by Bayer in 1983 and introduced in 1987. It became a blockbuster drug and sales reached two billion Euros in 2001. Now generics are introduced (after 2014). It is included in the essential medicinal list of WHO (2015).
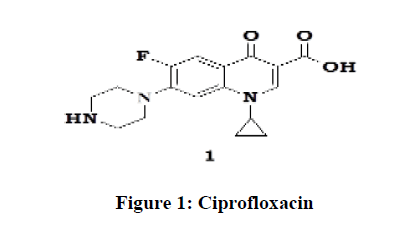
Figure 1: Ciprofloxacin
The original synthesis by Klaus Grohe [1] (Bayer) was starting from 2,4,5-trifluoro benzoyl chloride 2 and amino acrylate 3 condensation followed by cyclopropylamine reaction, cyclization and condensation with piperazine and finally salting with hydrochloride to produce the drug (Scheme 1).
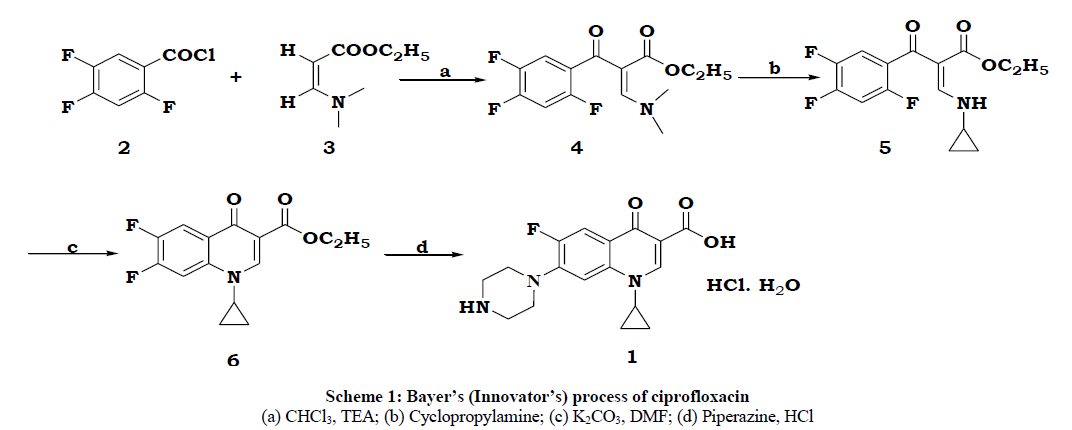
Scheme 1: Bayer’s (Innovator’s) process of ciprofloxacin
(a) CHCl3, TEA; (b) Cyclopropylamine; (c) K2CO3, DMF; (d) Piperazine, HCl
Later Bayer [2] team reported one pot synthesis of 3-quinoline carboxylic acid derivatives using the same raw materials to reduce the unit operations and increase the yields.
Bayer [3,4] group in another synthetic method started from 2,4-dichloro-5-fluorobenzoylchloride 7 is reacted with diethyl malonate in presence of magnesium ethoxide to give diester 8. The diester 8 is hydrolyzed and decaboxylated to give beta ketoester 9 and 9 was reacted with triethyorthoformate and acetic anhydride to give an enol ether 10. The enol ether compound 10 is reacted with cyclopropylamine to give cyclopropylamine derivative 11. This is cyclized with base to give cyclized product 12. This 12 was reacted with piperazine, hydrolyzed and salt formation led to the drug. Entire reaction process is given in Scheme 2.
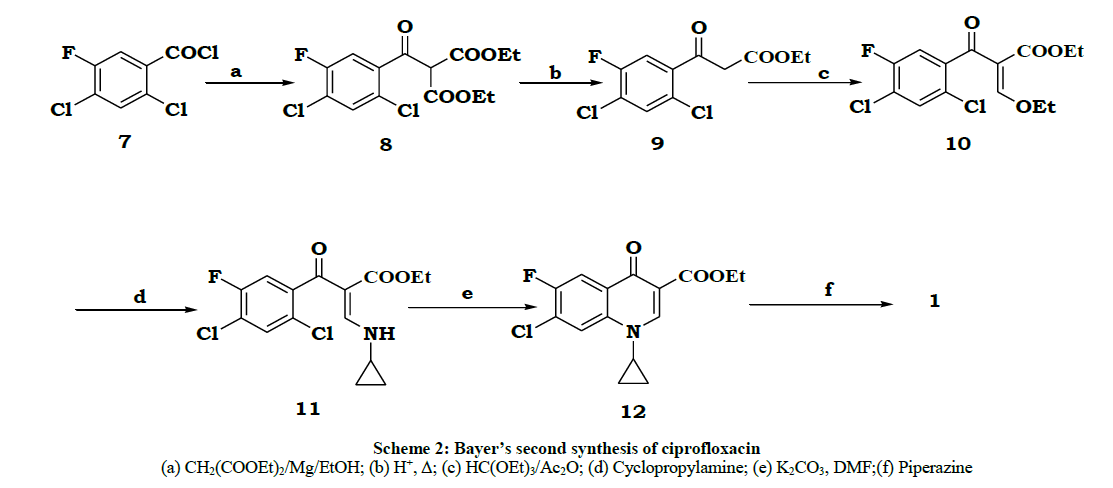
Scheme 2: Bayer’s second synthesis of ciprofloxacin
(a) CH2(COOEt)2/Mg/EtOH; (b) H+, Δ; (c) HC(OEt)3/Ac2O; (d) Cyclopropylamine; (e) K2CO3, DMF;(f) Piperazine
During 1992-1994, Natco developed a new process [5] for ciprofloxacin. In this process the conversion of compound beta ketoester to enol ether was modified. Instead of tri ethylothoformate and acetic anhydride reaction the beta ketoester was reacted with base (NaH) and then reacted with a complex prepared from dimethylformamide and dimethyl sulfate. The product formed in situ was reacted with cyclopropylamine to give 14 details are given in Scheme 3.
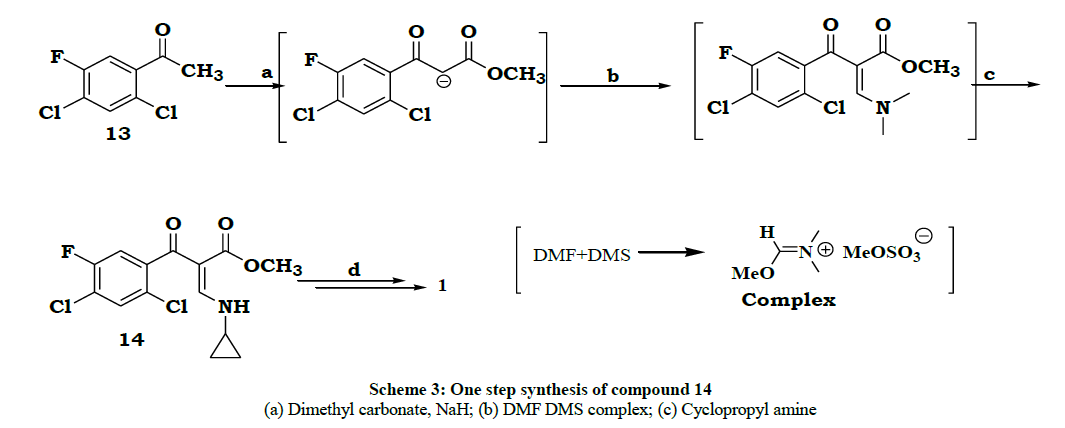
Scheme 3: One step synthesis of compound 14
(a) Dimethyl carbonate, NaH; (b) DMF DMS complex; (c) Cyclopropyl amine
In the year 2001, Natco [6] patented another process. In this process a nitro group was introduced to reduce the impurity formation during the final condensation with piperazine, Even though it was successful in bringing the selectivity, the removal of nitro group required two more steps, which industrially was not economical. The sequence of reactions are given in Scheme 4.
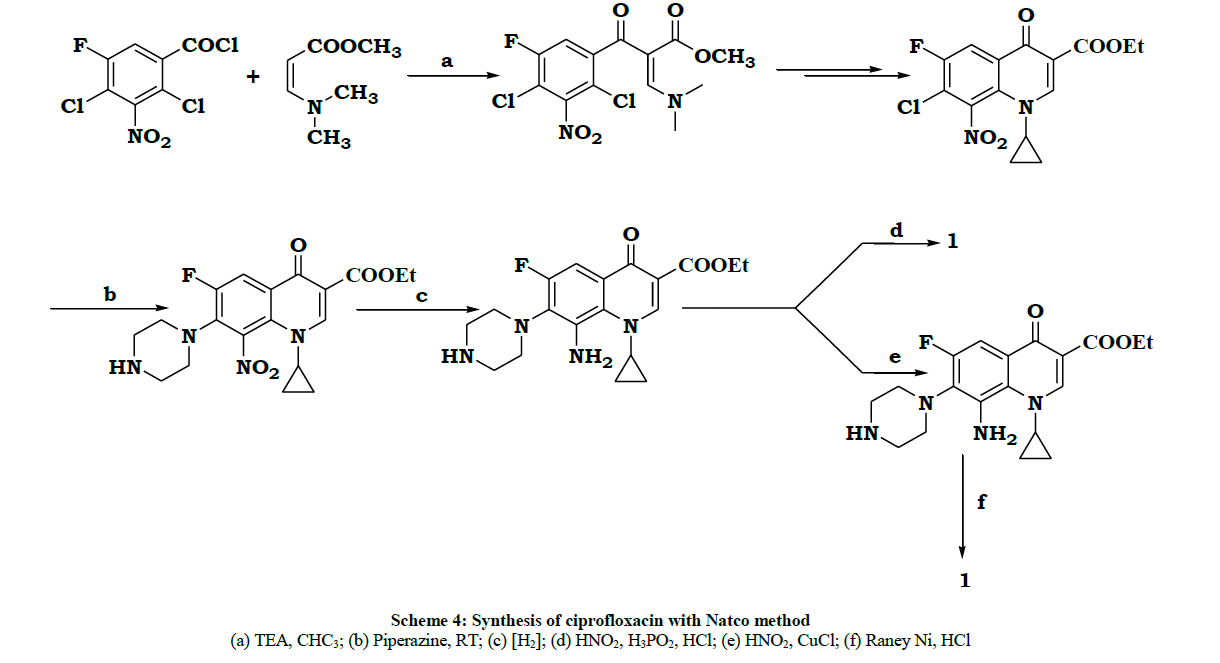
Scheme 4: Synthesis of ciprofloxacin with Natco method
(a) TEA, CHC3; (b) Piperazine, RT; (c) [H2]; (d) HNO2, H3PO2, HCl; (e) HNO2, CuCl; (f) Raney Ni, HCl
A number of other developments came during the years of 2001-2015, but most of them are minor improvements in yields. In 2012 Rao et al., [7] came with an improved process to reduce the unwanted fluoro substituted impurity 15.
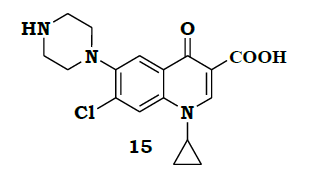
By adding Ferric Chloride (FeCl3) during piperazine condensation the minor impurity 15 was reduced to < 5%, which was removed during salt formation and crystallization process.
During 2010-2017 Suven [8] group developed a process for N-(3-Chloro-4-fluorophenyl)cyclopropylamine 16 to study Gould-Jacobs [9] method of preparation of ciprofloxacin. In that study they concluded that the process is industrially not feasible because of unwanted isomer compound 17 was formed ~45% (Scheme 5).
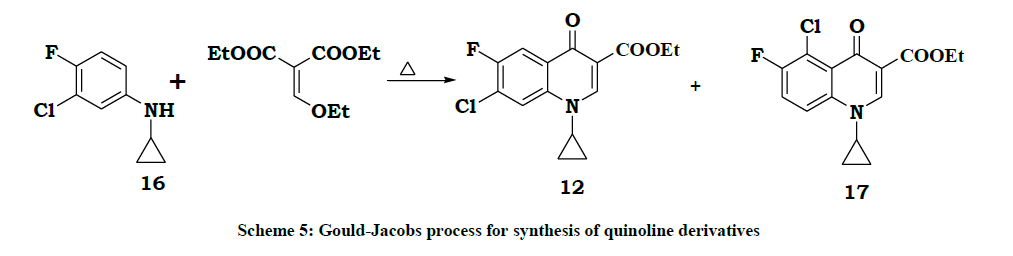
Scheme 5: Gould-Jacobs process for synthesis of quinoline derivatives
Flow chemistry
This chemistry was introduced about ~12 years ago in the synthesis laboratories [10]. This technology allows a continuous flow of reagent to be introduced at various points along a process stream, enabling interaction under highly controlled conditions. Flow system allow high through put chemistry to take place, often employing immobilized reagents or catalysts [11].
When compare to batch processes, flow process have minimal scale up problems. Instead of scaling up for mass production by increasing the size of the reactors, flow reactors can be scaled up by introducing more reactors in parallel while maintaining all of the parameters [12,13]. Carrying out flow reactions in lab has several advantages like increased temperature control, short residence times, easy of handling highly reactive substances and control of flow pressure in the process [14].
Jansen et al., [15] recently reported a continuous process based on Klaus Grohe (Bayer) method and they claimed the overall yield of eight step sequence as 60%.
Materials and Methods
Most of the reagents used in this work were obtained from commercial suppliers and were of LR/AR grade. Solvents were purified before use by standard procedures. Melting points were determined using open capillary tubes on POLMON melting points apparatus (Model-96) and are uncorrected. 1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded by using a Bruker 400 Spectrometer with Tetramethylsilane (TMS) as internal standard. IR spectra were recorded on a Perkin-Elmer Spectrum 100 FTIR Spectrophotometer as KBr pellets or with the neat products. Mass spectra were recorded on an API 2000 LCMS/MS Applied Bio Systems MDS Sciex spectrometer. Microanalysis was performed on a Perkin-Elmer 240CHN elemental analyzer. Analytical TLC was conducted on E-Merck 60F254 aluminium-packed plates of silica gel (0.2 mm). Developed plates were visualized by using UV light or in an iodine chamber. High Performance Liquid Chromatography (HPLC) was performed by using a Shimadzu 2010 instrument.
General procedure for synthesis of 7-Chloro-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid methyl ester (19)
To a solution of 3-dimethylamino-acrylic acid methyl ester5b (62.3 g, 0.48 mol) and toluene (400 ml), Triethylaluminum (TEA) (66.5 g, 0.65 mol) was added at 10-15° for 15 min after completion of the addition stirred at 10-15°C. After 15 min was added a solution of 2,4-dichloro 5- fluoro benzoylchloride (100 g, 0.43 mol) in toluene (200 ml) at 10-15°C for 30 min and stirred for 10 min at RT, slowly reaction mass temperature raised to 80-85°C. The mixture was maintained at 80-85°C for 4 h. After completion of reaction (Monitored by TLC) reaction mixture was cooled to RT and cyclopropylamine (28.0 g, 0.48 mol) was added to the reaction mixture at RT over 15 min. The mixture was stirred at RT for 1 h. After completion of the reaction (Monitored by TLC) to the reaction mixture added potassium carbonate (66.0 g, 0.48 mol) and DMF (400 ml) at RT and stirred for 10 min. After 10 min reaction mixture temperature raised to 120-125° (at 110-115°C toluene was collected) and stirred at 120-125°C for 4 h. After completion of the reaction (Monitored by TLC) reaction mass was cooled to 35-40°C and quenched into ice water (2.0 liter) and stirred at RT. After 1.5 h filtered the solid and washed with water (200 ml) and hexane (500 ml) to get 19 (85.0 g), Yield 65%. Product 19 was confirmed on the basis of spectral data.
Description: off white color solid; M.p: 240.2-243.3°C; IR (in KBr, cm-1): 3436, 2952, 1701, 1731, 1613, 1639, 1478, 1196, 803; 1H-NMR (CDCl3, 400 MHz) (δ ppm): 8.61 (s, 1H), 8.23 (d, J=8.9 Hz, 1H), 8.0 (d, J=5.8 Hz, 1H), 3.9 (s, 3H), 3.48-3.44 (m, 1H), 1.41-1.36 (q, 2H), 1.18- 1.14 (q, 2H); 13C-NMR (CDCl3, 100 MHz) (δ ppm): 172.72, 165.89, 156.98, 154.49, 149.0, 137.17, 128.74, 128.68, 126.94, 118.95, 114.0, 113.82, 110.50, 525.24, 34.79, 8.28; Mass for C14H11ClFNO3 [M+1], found 297.7.
Result and Discussion
In this paper we wish to report a process similar to Klaus Grohe (Bayer) [1], but the starting material is different, and is 2,4-dichloro-5-fluoro benzoyl chloride 7. A telescopic single step process is developed for the key intermediate 19, with an overall yield of 65% and this process is commercially viable. From 19 in one step [7] the drug can be synthesized. The whole process is described in Scheme 6.
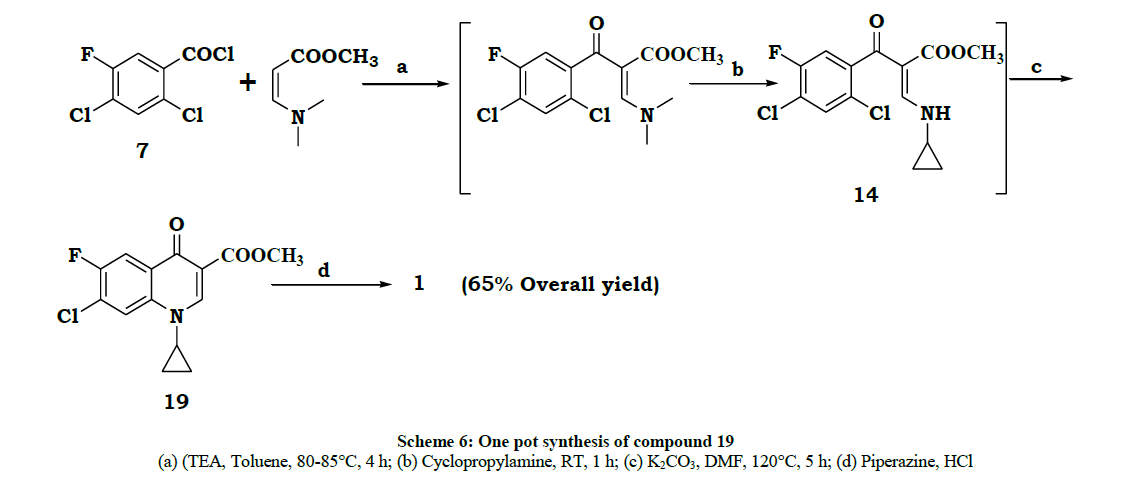
Scheme 6: One pot synthesis of compound 19
(a) (TEA, Toluene, 80-85°C, 4 h; (b) Cyclopropylamine, RT, 1 h; (c) K2CO3, DMF, 120°C, 5 h; (d) Piperazine, HCl
Conclusion
We have developed a two-step synthesis of ciprofloxacin in best possible yields currently.
Acknowledgement
We would like to thank Suven life sciences for providing excellent analytical facilities and our CEO Mr Jasti for allowing us to publish this work.
References
- K. Grohe, H. Heitger, Libig Ann. Chem., 1987, 29.
- R. Zerbes, P. Naab, G. Frankowiak, H. Diehl, (Patent-US 5,639,886A), 1997.
- K. Grohe, H.J. Zeiler, K. Metzger, (Patent-DE 3142854), 1983.
- K. Grohe, H.J. Zeiler, K. Metzger, (Patent-US 4670444), 1987.
- A. Veerareddy, M. Pullareddy, Indian Patent, 1999.
- P.R. Muddasani, V.C. Nannapaneni, (Patent-WO 2001085692A2), 2001.
- D.R. Rao, P. Ravi, B. Sunil, A.P. Kumat, M. Sunil, (Patent-WO 2012127505), 2012.
- V.R. Arava, S.R. Bandatmakur, Synthesis., 2013, 45, 1039-1044.
- R.G. Gould, W.A. Jacobs, J. Am. Chem. Soc., 1939, 61(10), 2890.
- A. Kirshning, Beilstein. J. Org. Chem.,2009, 5, 15.
- T. Schwallbe, D. Kadiziminsz, G. Jas, QSAR Combi. Sci., 2005, 24.
- P. Styring, A. Parracho, Beilstein. J. Org. Chem.,2009, 5(18).
- Y. Yamada, K. Tori, Y. Uozumi, Beilstein. J. Org. Chem.,2009,5(18), 47.
- J. Brandt, T. Wirth, Beilstein. J. Org. Chem.,2009,5, 30.
- H. Lin, C. Dai, T.F. Jamison, K.F. Jensen, Angew. Chem. Int. Ed., 2017, 56, 8870-8873.



