Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 4
Computational Modeling and Simulation of Toxin-antitoxin System in Acinetobacter baumannii: HicB as a Dynamic Factor for Therapeutic Target
Maryam Pourhajibagher1, Nava Hosseini2 and Abbas Bahador1,3*
1Dental Research Center, Dentistry Research Institute, Tehran University of Medical Sciences, Tehran, Iran
2Department of Microbiology, Faculty of Biology, College of Science, University of Tehran, Tehran, Iran
3Department of Microbiology, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran
- *Corresponding Author:
- Abbas Bahador
Dental Research Center, Dentistry Research Institute
Tehran University of Medical Sciences
Tehran, Iran
Abstract
Acinetobacter baumannii is a ubiquitous Gram-negative bacillus bacterium that is frequently found in the nosocomial infections. Regard to the rapid development of resistance against various antimicrobial agents due to the high ability of natural genetic transformation, A. baumannii is resistance to many of the antibiotics. Toxin-antitoxin system is one of the virulence agents in A. baumannii that can be involved in the emergence of drug resistance. HicB as a toxin system is an ideal target for the design of new classes of antimicrobial agents against A. baumannii. The goals of this work was to model the complete tridimensional structure of the HicB protein using computational methods and to assess structural and functional of it. The consistency of modeled structure was confirmed by different servers to high accuracy for modeling and dynamics simulation. The molecular dynamic simulation and virtual screening results indicate that HicB is a stable protein from A. baumannii. These results provide new insights for the molecular understanding and development of new antibacterial drugs against toxin system involved in A. baumannii pathogenesis.
Keywords
Acinetobacter baumannii, Bioinformatics, Drug target, In silico, Toxin-antitoxin system
Introduction
Nowadays, nosocomial infections caused by Acinetobacter baumannii are widespread problem in all countries [1]. A. baumannii develops resistance against antibiotics which is mainly due to less susceptible nature of this organism. Immunocompromised patients are susceptible to be affected by opportunistic infections caused by A. baumannii [2]. The occurrence of multi-drug resistance strains of A. baumannii has evidently limited the therapeutic procedures, so it’s logical to determine virulence factors determining the pathogenicity of A. baumannii. This organism is capable to grow on both biotic and abiotic surfaces and is able to survive in unfavorable conditions [1]. One of the main reasons of A. baumannii long-period survival is due to the presence of dormant cells in A. baumannii infections [3]. Toxin-antitoxin modules are attributed to play a key role in different cell processes, like stress management or survival of bacteria towards inappropriate conditions like forming persister cells [4-6].
HicBA is one of the type II toxin-antitoxin modules which first identified in Haemophilus influenza as a bicistronic locus which is related closely to the pilus gene cluster [5]. Also, it has been proved that starvation conditions leads to up-regulation of HicBA system. Recently this system has been studied in some bacteria. HicA toxin in Escherichia coli inhibits the cell growth by mRNA degradation. In Burkholdaria pseudomallei and Pseudomonas aeruginosa, HicA toxin induces growth arrest and persistence in the presence of antibiotics, like ciprofloxacin or ceftazidime [7]. The persistence phenomenon towards ciprofloxacin or ceftazidime antibiotics can be modulated differentially depending on the level induction of the HicA toxin expression. The deletion of HicBA locus from P. pseudomallei K96243, the frequency of persister cells reduced specially after ciprofloxacin exposure. In structural aspect, HicA toxin forms double strand-binding domain-like fold, which is composed of triple-stranded B-sheet with two helices [8], but HicB antitoxin has DNA-binding domain which is fused to a degraded RNase H fold. This system is encoded differently from other toxin-antitoxin modules, because toxin is encoded before antitoxin [9]. By knowing this, it can be proposed that HicB is an ideal target for the design of new classes of antimicrobial agents against A. baumannii.
In this scenario, the computational approach of bioinformatics comes as a great ally of experimental methodology. Computational- or in silico methods are based on algorithms that can make predictions with a variety of purposes, such as predicting the structure of HicB as a virulence factor in A. baumannii. In this work, computational biology methods were applied, following the methodology previously described by our group [10-14] to an in silico analysis of HicB protein, which has been described as a drug target for antimicrobial therapy.
Materials and Methods
Sequence and natural variants retrieval
The primary sequence of antitoxin HicB (accession number: KZA08420.1) of A. baumannii was retrieved in FASTA format from the Universal Protein Resource (UniProt) (http://www.uniprot.org/). The online protein Basic Local Aligment Search Tool (BLAST) of HicB sequence was performed to identify the known homologous tridimensional structure which can be further used as a template to model HicB.
Structural modeling
Secondary structure of our protein under observation was predicted using European Bioinformatics Institute (EMBL-EBI; https://www.ebi.ac.uk/services). The tridimensional structures of HicB were created based on comparative and Ab Initio modeling. The generated structures were then structurally aligned to the crystallographic structure of HicB (PDB ID: 4P78) and interactions of HicB molecule were investigated.
Validation of modeled structure
The quality of the model with lowest DOPE score was then validated using PROCHECK (www.ebi.ac.uk/thornton-srv/PROCHECK/), ERRAT (http://services.mbi.ucla.edu/ERRAT), VERIFY3D (http://services.mbi.ucla.edu/Verify_3D/) program available at SAVES server, and QMEAN (https://swissmodel.expasy.org/qmean/).
In addition, ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php) was used for calculating the native conformation and Ramachandran plots generated using Rampage (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php).
Analysis tools for determination of physicochemical properties including Isoelectric Point/Molecular Weight (pI/Mw), a tool for predicting HicB (pI) and MW, ProtScale for amino acid scale representation such as hydrophobicity plots, ProtParam to assess numerous physicochemical parameters, peptide mass as a tool for theoretically cleaving HicB and calculating the masses of its peptide, and any known processing of cellular or post-translational modifications, were used from the tools page available at UniProt (http://expasy.org) and Expasy ProtParam server (http://www.expasy.org/cgi-bin/protpraram).
The propensities of individual and the conditional probability amino acids to form particular secondary structures were demonstrated using the Garnier-Osguthorpe-Robson (GOR) IV method (https://npsa-prabi.ibcp.fr). The ligand binding site, enzyme commission (EC) numbers, Predicted normalized B-factor, and active sites were found by the ITASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER).
Prediction of intrinsic disorder
Protein disordered prediction is important in determining protein function and protein folding pathway. In order to identify segments of disordered and higher flexibility, the tools DisEMBL (http://dis.embl.de/), Globplot (http://globplot.embl.de/), PRDOS (http://prdos.hgc.jp/cgi-bin/ top.cgi) and meta PRDOS (http://prdos.hgc.jp/cgi-bin/meta/top.cgi) were used.
Molecular dynamics
The stability and conformational changes in the native and the mutants were assessed through the analysis of Root-mean-square deviation (RMSD).
The phylogenetic analysis was performed using the ConSurf server (http://consurf.tau.ac.il). It determined the evolutionary conservation degree of each HicB amino acid. As well as, Absolute surface area (ASA) (http://www.abren.net/asaview/) was used to provide the graphical representation of solvent accessibility of amino acid residues in proteins, with known structures. Proteins structures have been taken from the Protein Data Bank and DSSP program was used to compute their solvent accessibility. Difference Distance Map (DDM) (http://pdbflex.org/cluster.html) was used to show the changes of distances between equivalent C-alpha atoms in the two structures with maximum structural flexibility.
Sequence retrieval
NCBI GenBank database showed that HicB has 137 amino acids. By BLAST, similarity search against HicB identified in A. baumannii displayed that it is similar to the protein structure with accession number of 4P78. The total score for protein alignment between HicB and 4P78 was 71.2, with 44% and 21% query cover and identity, respectively.
The protein has 135 amino acids with molecular weight of 15246.42 and estimated theoretical isoelectric point (pI) of 5.02. The Extension coefficient of protein is 7450 indicates that at a definite wavelength how much light was absorbs by a protein. The instability index (II) is 38.86 that displaying protein stability. The instability index (II) of HicB was near to 40. Instability index smaller than 40 for a protein is expected as stable. Protein may be unstable with instability index more than 40.
On the other hand, the hydrophilic nature is predicted by the grand average of hydropathy (GRAVY) index value of -0.196. The thermostability of globular proteins is calculated by aliphatic index. The Aliphatic index of HicB is 81.04 indicating the relative volume of aliphatic side chains (Leucine, valine, isoleucine and alanine).
Prediction of HicB structures
Initial structure prediction showed that HicB had 15 positively charged residues (Arg + Lys) and 20 negatively charged residues (Asp + Glu) in A. baumannii. The amino acid compositions of HicB are: Ala (A) 8.9%, Arg (R) 3.7%, Asn (N) 5.2%, Asp (D) 6.7%, Cys (C) 0.7%, Gln (Q) 1.5%, Glu (E) 8.1%, Gly (G) 6.7%, His (H) 1.5%, Ile (I) 9.6%, Leu (L) 6.7%, Lys (K) 7.4%, Met (M) 2.2%, Phe (F) 9.6%, Pro (P) 6.7%, Ser (S) 4.4%, Thr (T) 3.7%, Trp (W) 0.0%, Tyr (Y) 3.7%, Val (V) 3.0%, Pyl (O) 0.0% and Sec (U) 0.0%. The N-terminal of the sequence considered is M (Met). The estimated half-life is: 30 h (Mammalian reticulocytes, in vitro), >20 h (yeast, in vivo) and >10 h (E. coli, in vivo).
As you see in Figure 1, prediction of secondary structure ensures whether amino acids lie in strand, helix or coil. The inspection of GOR IV analysis in Table 1 show that alpha helix dominates over random coil, followed by extended strand.
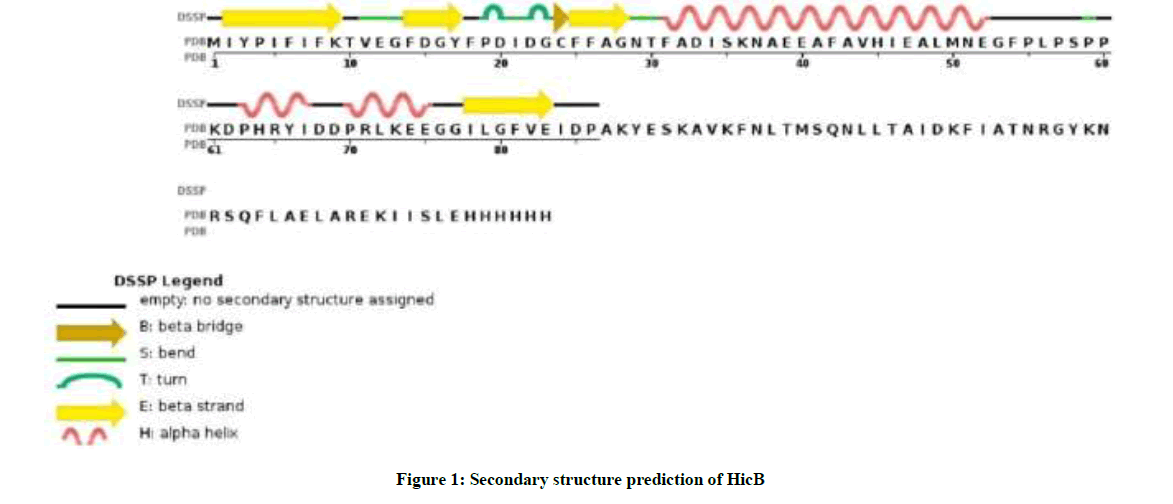
Figure 1: Secondary structure prediction of HicB
| Groups | Prevalence |
|---|---|
| Alpha helix | 77 is 56.2% |
| 310 helix | 0 is 0.00% |
| Pi helix | 0 is 0.00% |
| Beta bridge | 0 is 0.00% |
| Extended strand | 13 is 9.49% |
| Beta turn | 0 is 0.00% |
| Bend region | 0 is 0.00% |
| Random coil | 47 is 34.31% |
Table 1: The prevalence of different structure in HicB
4P78 has 4 unique types of molecules in this entry. The entry contains 2507 atoms, of which 0 is hydrogen and 0 is deuterium. Molecule 1 is a protein called HicB3 antitoxin, Molecule 2 is a protein called HicA3 Toxin, Molecule 3 is GLYCEROL (Three-letter code: Gol) (Formula: C3H8O3) and Molecule 4 is water (Figure 2).
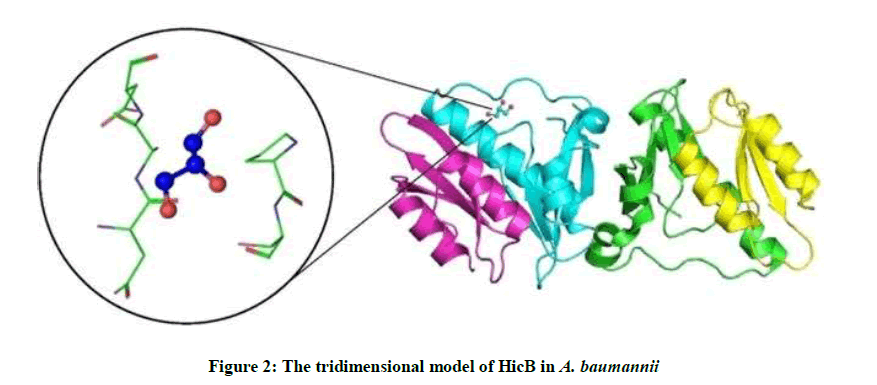
Figure 2: The tridimensional model of HicB in A. baumannii
As well as, the sites of other amino acids that are surrounded the Gol is shown in Figure 3.
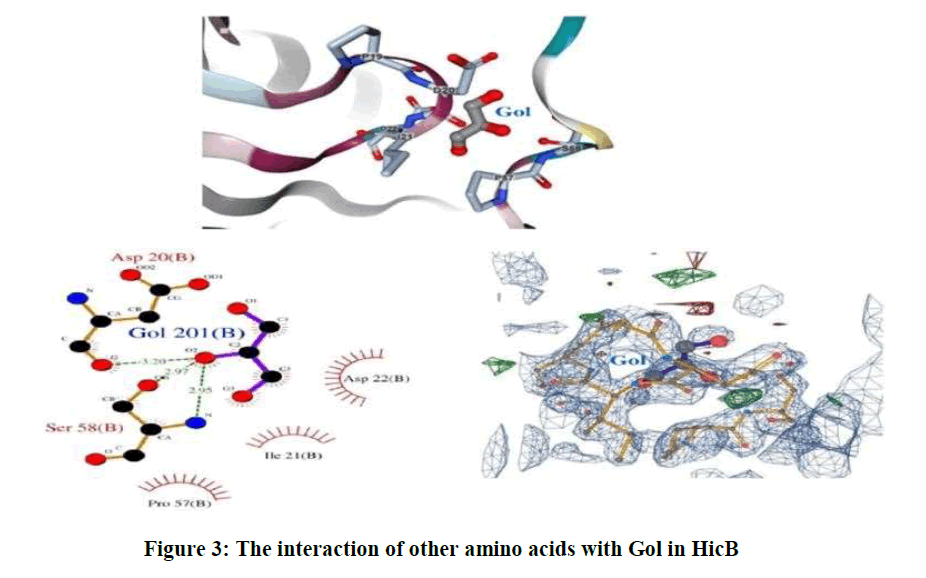
Figure 3: The interaction of other amino acids with Gol in HicB
Structure validation
The QMEAN Z-score measures the absolute quality of a model by comparing its QMEAN score to scores of experimentally solved proteins. The final model obtained a QMEAN Z-score of <1, which falls within the range of scores found for reference structures of the same size (Figure 4A). A similar analysis is made by ProSA web algorithm, according to which the final structure obtained a Z-score of -6. 1 and therefore also falls within the range of scores found on similar sized proteins, with an X-ray quality (Figure 4B). According to the results of ERRAT in Figure 4C, on the error axis, two lines are drawn to indicate the confidence with which it is possible to reject regions that exceed that error value. Expresses as the percentage of the protein for which the calculated error value falls below the 95% rejection limit. Good high resolution structures generally produce values around 95% or high. For lower resolutions (2.5 to 3A) the average overall quality factor is around 91%.
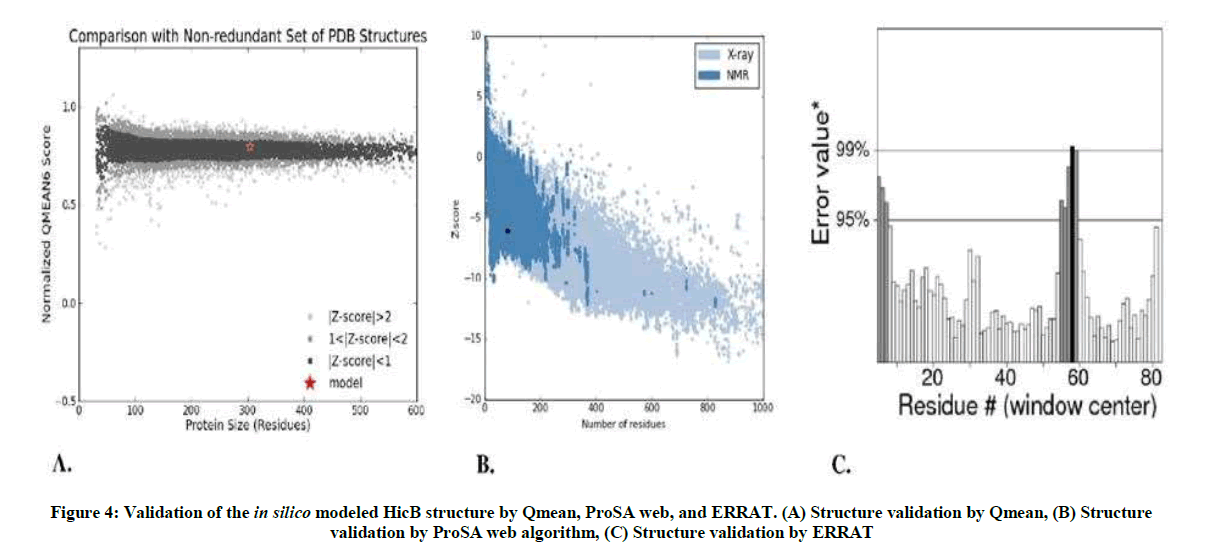
Figure 4: Validation of the in silico modeled HicB structure by Qmean, ProSA web, and ERRAT. (A) Structure validation by Qmean, (B) Structure validation by ProSA web algorithm, (C) Structure validation by ERRAT
On the Ramachandran plot generated by Rampage, the final model had 95.7% residues in favored regions, 2.7% in allowed regions and 1.6% in outlier regions (Figure 5).
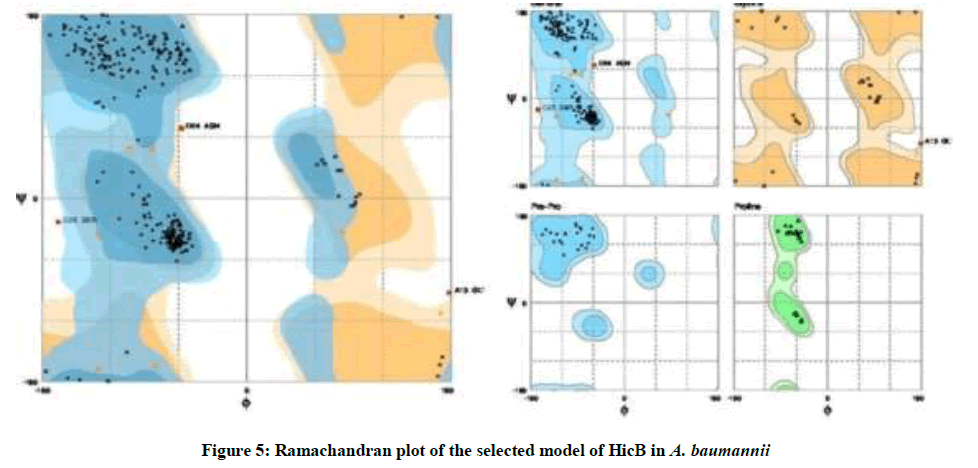
Figure 5: Ramachandran plot of the selected model of HicB in A. baumannii
After the analysis performed at the I-TASSER server, it was found that ligand binding site residues were 33, 34, and 37 amino acids with 0.10 C-score. In addition, the active site residue was located at 135 residue with 0.495 TM-score, 0.154 C-score, 4.54 RMSD, and 0.053 percentage sequence identity in the structurally aligned region.
Prediction of intrinsic disorder
Protein disorder can be defined as the region without regular secondary structure and of high degree of flexibility in protein chain. The sequence was submitted to different online server having free access and the result of different server is given in Figure 6A-D.
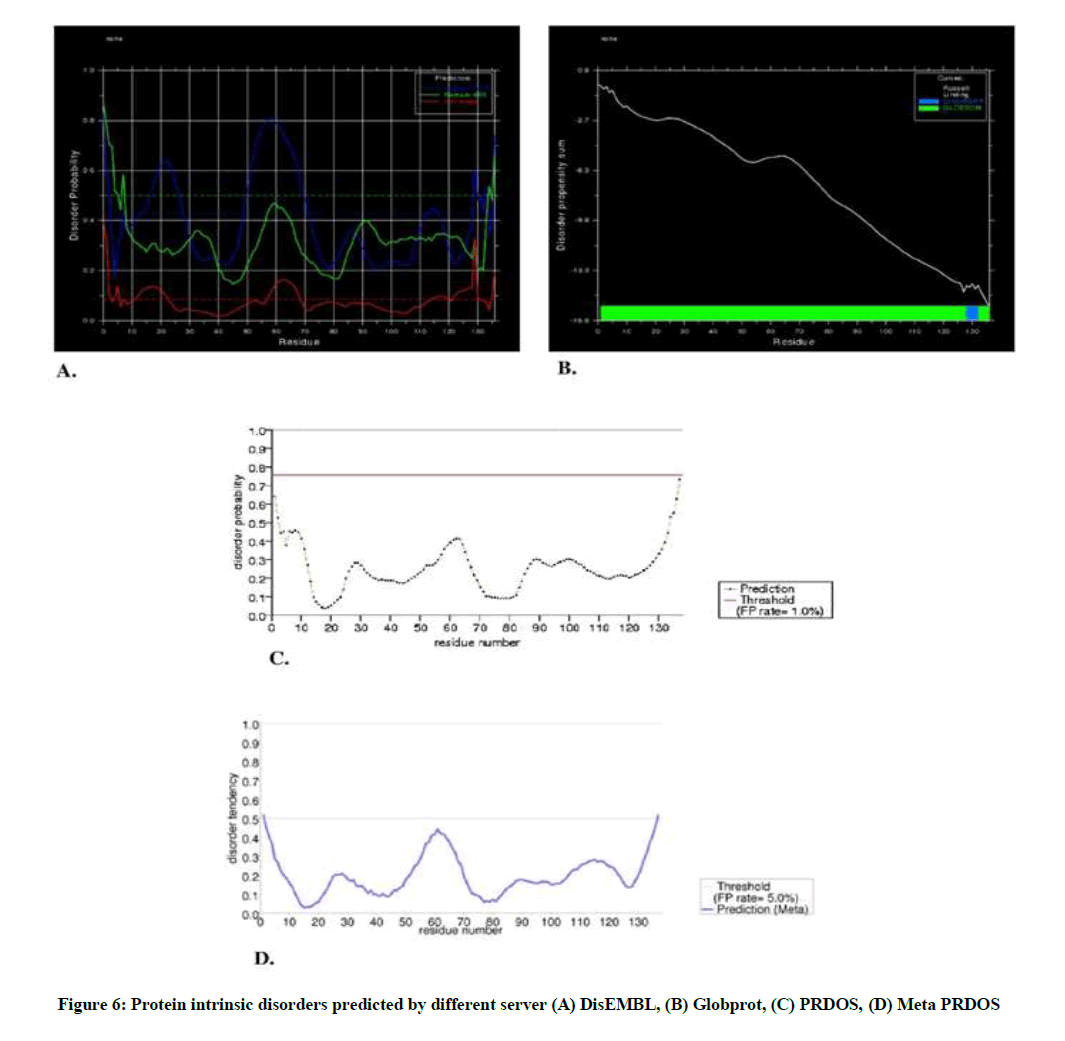
Figure 6: Protein intrinsic disorders predicted by different server (A) DisEMBL, (B) Globprot, (C) PRDOS, (D) Meta PRDOS
In DisEMBL server, disordered by Loops/coils definition were 15-28, 50-72, 130-137; disordered by Hot-loops definition were 13-23, 59-69, 123-131; whereas there was no disordered by Remark-465 definition. On the other hand, in Globprot server, disordered by Russell/Linding definition were 128-132 and Potential globular domains (GlobDoms) by Russell/Linding definition were 1-137.
Molecular dynamics simulation
Molecular dynamics simulation solves the classical equation of motion. The RMSD of backbone atoms as a function of time was evaluated. The ASA analysis assesses the surface area of the protein that is accessible to the solvent, where high ASA values suggest relative expansion. As seen in Figure 7, color scheme used is: Blue -> Positive charged residues (R, K, H), Red -> Negative charged residues (D, E), Green-> Polar uncharged residues (G, N, Y, Q, S, T, W), Yellow -> Cystein, and Gray -> Hydrophobic residues (All others).
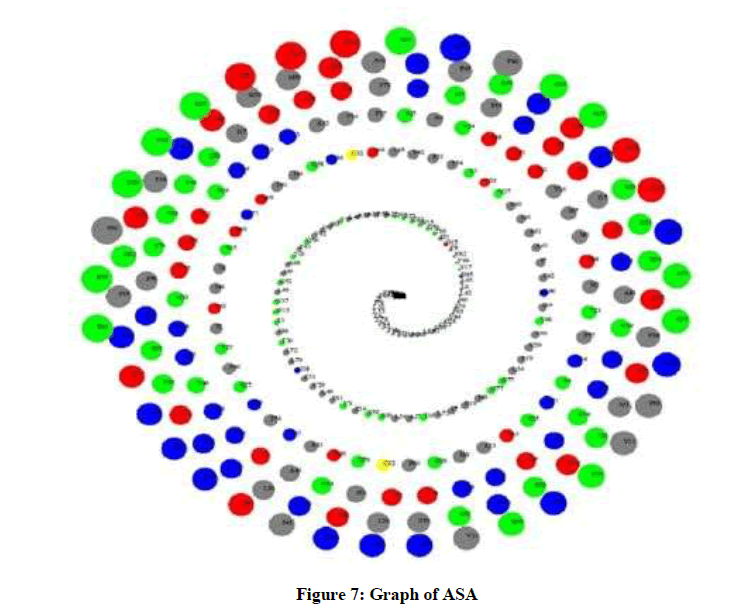
Figure 7: Graph of ASA
Based on the results of DDM, distance range is -31.2 to 20.7 angstroms. Red color indicates an increase and blue indicates a decrease of the distance between C-alpha atoms. X and Y-axes correspond to aligned sequences of two structures. The intensity of the color describes the magnitude of change (Intense red means large increase of the distance, pale blue means small decrease etc.). Square blocks of low color intensity located on the matrix diagonal correspond to rigid domains and subdomains. The stripes of high color intensity between on-diagonal blocks of low color intensity indicate rearrangements of rigid regions connected by flexible linkers (Figure 8).
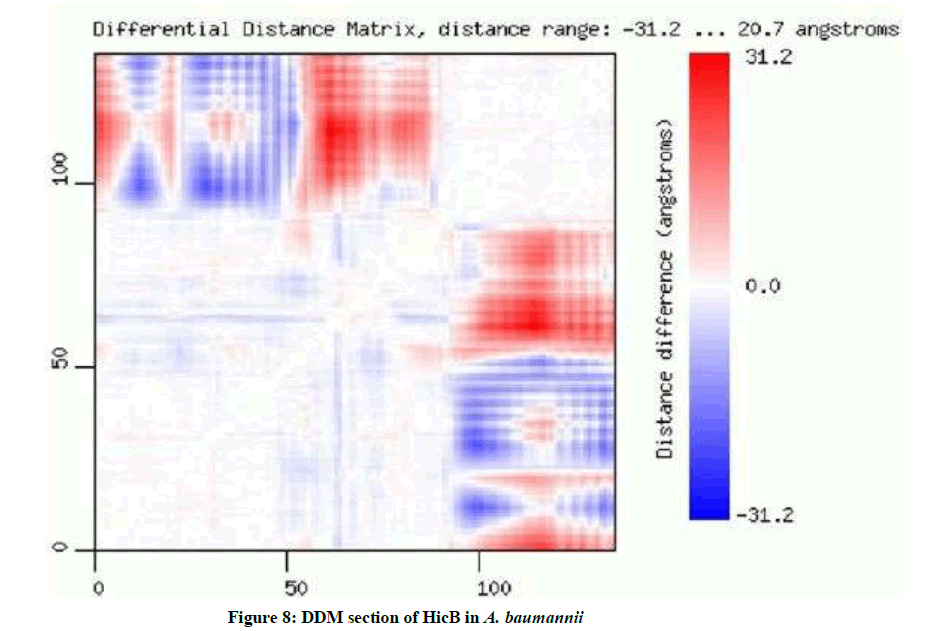
Figure 8: DDM section of HicB in A. baumannii
Conservation analysis
ConSurf server is a bioinformatics tool that estimates the evolutionary conservation of amino acids in a protein and projects the scores on their molecular surface, using a coloring scheme. Important amino acids are usually strongly conserved throughout evolution. As you see in Figure 9, highly conserved positions are colored bordeaux, intermediately conserved positions are colored white and variable positions are colored turquoise.
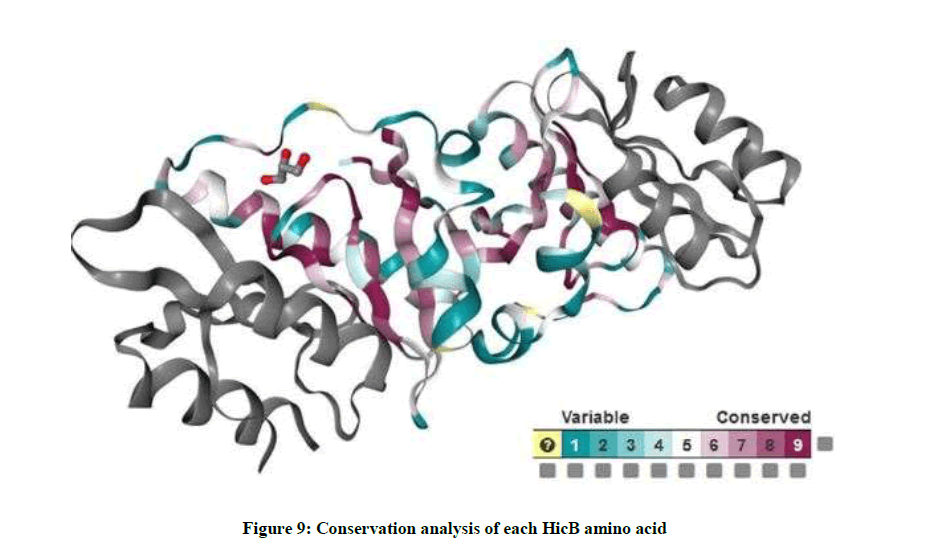
Figure 9: Conservation analysis of each HicB amino acid
Conclusion
We have successfully modeled the complete tridimensional structure of HicB as a toxin in A. baumannii. It is important to note that the in silico analysis is an approach that is able to introduce the protein structure which enables the understanding of mechanism of protein’s function. We modeled for the first time, the HicB toxin as a stable protein from A. baumannii which has a role in pathogenesis and can be a good drug target for pathogenic inhibition.
References
- M. Pourhajibagher, M. Mokhtaran, D. Esmaeili, A. Bahador, Der Pharmacia Lettre, 2016, 8, 225-229.
- Z. Farshadzadeh, F.B. Hashemi, S. Rahimi, B. Pourakbari, D. Esmaeili, M.A. Haghighi, Front. Microbiol., 2015, 6, 1146-1153.
- M. Pourhajibagher, F.B. Hashemi, B. Pourakbari, M. Aziemzadeh, A. Bahador, Open Microbiol. J., 2016, 10, 32-42.
- M. Beheshti, M. Talebi, A. Ardebili, A. Bahador, A.R. Lari, J. Pharm. Bioallied. Sci., 2014, 6, 229-32.
- M. Pourhajibagher, M. Mokhtaran, D. Esmaeili, A. Bahador. Der Pharma Chemica, 2016, 8, 347-351.
- V. Tsilibaris, G. Maenhaut-Michel, N. Mine, L. Van Melderen, J.Bacteriol, 2007, 189, 6101-6108.
- A. Butt, V.A. Higman, C. Williams, M.P. Crump, C.M. Hemsley, N. Harmer, Biochem J, 2014, 459, 333-344.
- M. Safari, A.S. Mozaffari Nejad, A. Bahador, R. Jafari, M.Y.Alikhani, Saudi J. Biol. Sci., 2015, 22, 424-429.
- M. Muthuramalingam, J.C. White, C.R. Bourne, Toxins (Basel)., 2016, 8, e214.
- M. Pourhajibagher, A. Bahador, Drug Research., 2016; 66:1-5.
- M. Pourhajibagher, A. Bahador, Photodiagnosis Photodyn. Ther., 2016, 16, 154-160.
- M. Pourhajibagher, A. Bahador, Photodiagnosis Photodyn. Ther., 2017, 17, 98-102.
- M. Pourhajibagher, S. Gharesi, R. Ghorbanzadeh, Der Pharma Chemica., 2015, 7, 243-250.
- M. Pourhajibagher, M.M. Karimi Yazi, A. Bahador, Der Pharma Chemica., 2015, 7, 301-306.



