Research Article - Der Pharma Chemica ( 2019) Volume 11, Issue 2
Design, Efficient Synthesis and Antimicrobial Evaluation of Some Novel Pyrano[2, 3-b][1, 8]Naphthyridine and Pyrrolo [2,3-f][1,8] Naphth- yridine Derivatives
Nadia Ali Ahmed Elkanzi1,2*, Amira A Ghoneim1,3 and Rania B Bakr4,52Department of Chemistry, Faculty of Science, Aswan University, P.O. box 81528, Aswan, Egypt
3Department of Chemistry, Faculty of Science, Zagazig University, Zagazig, Egypt
4Department of Pharmaceutical Chemistry, College of Pharmacy, Jouf University, Sakaka, Aljouf 2014, Kingdom of Saudi Arabia
5Department of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Beni Suef University, Beni-Suef 62514, Egypt
Nadia Ali Ahmed Elkanzi, Department of Chemistry, College of Science, Jouf University, P.O. Box: 2014, Sakaka, Saudi Arabia, Email: eduj@ju.edu.sa
Abstract
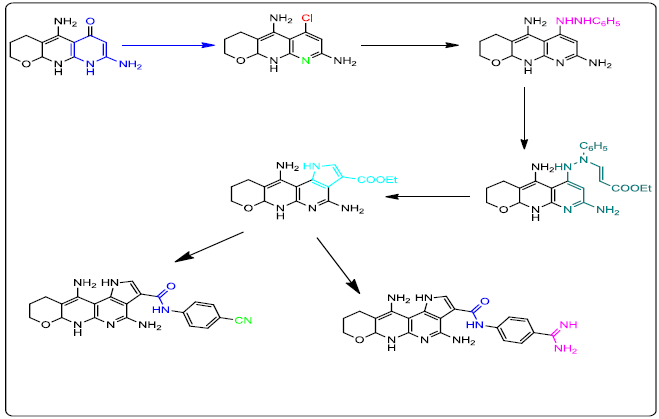
Reaction of Ethyl 5,7-diamino-3,4,8, 8a-tetrahydro-2H-pyrano[2, 3-b]pyridine-6-carboxylate (5) with acetonitrile and acetic acid afforded the corresponding 5,8-diamino-3,4,10,10a-tetrahydro-2H-pyrano[2,3-b][1,8]naphthyridin-6(9H)-one (6) which was reacted with phosphorus ox chloride to give crude 7. A solution of crude 7 was added to phenyl hydrazine to afford 6-(2-phenylhydrazinyl)-3,4,10,10a-tetrahydro-2Hpyrano[ 2,3-b][1,8]naphthyridine-5,8-diamine (8). Treatment of compound 8 with ethyl propiolate yielded compound 9 which was heated under reflux to produce the corresponding ethyl 4,11-diamino-1,6,6a,8,9,10-hexahydropyrano[2,3-b]pyrrolo[2,3-f][1,8]naphthyridine-3- carboxylate (10). Hydrolysis of the latter compound 10 with 10% sodium hydroxide afforded the acid derivative 11 which was reacted with 4- aminobenzamidinedihydrochloride (12), benzotriazol-1-yloxytris (pyrrolidino) phosphonium-hexafluorophosphate andN-ethyldiisopropylamine to afford the target compound 13. The formation of 4,11-diamino-N-(4-cyanophenyl)-1,6,6a,8,9,10-hexa-hydropyrano[2,3- b]pyrrolo[2,3-f][1,8]naphthyridine-3-carboxamide 15 was achieved by reaction of benzotriazol-1-yloxytris (pyrrolidino) phosphoniumhexafluorophosphate, 4-cyanoaniline (14) and N-ethyldiisopropylamine with the key intermediate (11). The structure of all the new compounds was demonstrated by elemental analysis, Infrared (IR), Proton Nuclear Magnetic Resonance (1H-NMR) and Carbon-13 Nuclear Magnetic Resonance (13C-NMR) spectra and mass spectra. Antimicrobial activity of these compounds 5-8, 10, 11, 13 and 15 was evaluated against Bacillus subtilis, Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa bacteria Aspergillus flavus and Candida albicans. 5,8- Diamino-3,4,10,10a-tetrahydro-2H-pyrano[2,3-b][1,8]naphthyridin-6(9H)-one (6) was found to be the most active against all the tested microorganisms. Furthermore, molecular modeling study had been performed on the target compound (6) to expect the mode of action of this candidate as a lead for designing other antimicrobial agents.
Keywords
Naphthyridines, Pyranoheterocycles, Pyridine derivatives, Quinoline derivatives, Molecular docking.
Introduction
The chemistry of naphthyridine derivatives had received much attention due to its diverse biological properties including antioxidants [1,2], anticancer [3,4], and anti-inflammatory activity [5,6], antitubercular [7], antiallergic [8] and antimicrobial agents [9-11]. For example, the naphthyridine derivative (1) (Figure 1) exhibited significant antimicrobial activity towards Escherichia Coli in comparison with the standard drug streptomycin at concentration=400 μg/disc[12]. In addition, pyrano heterocyclic revealed great importance due to their broad field of biological activities as enzyme substrates [13,14], and widely used as antimicrobial agent [15,16]. For example, Al-Saleem et al. [17] prepared 5,7-dibromo-3-ethoxycarbonyl-8-hydroxy-2-oxo-2H-1-benzopyrane (2) (Figure 1) exhibited comparable antifungal activities to that shown by fluconazole drug as reference standard. Finally, pyrrole moiety was known to show excellent antimicrobial activity [18,19], for example, the pyrrolo derivative (3) was evaluated for antimicrobial activity, it showed two times more activity (MIC=256 μg/ml) than fluconazole (MIC=512 μg/ml) as antifungal [20].
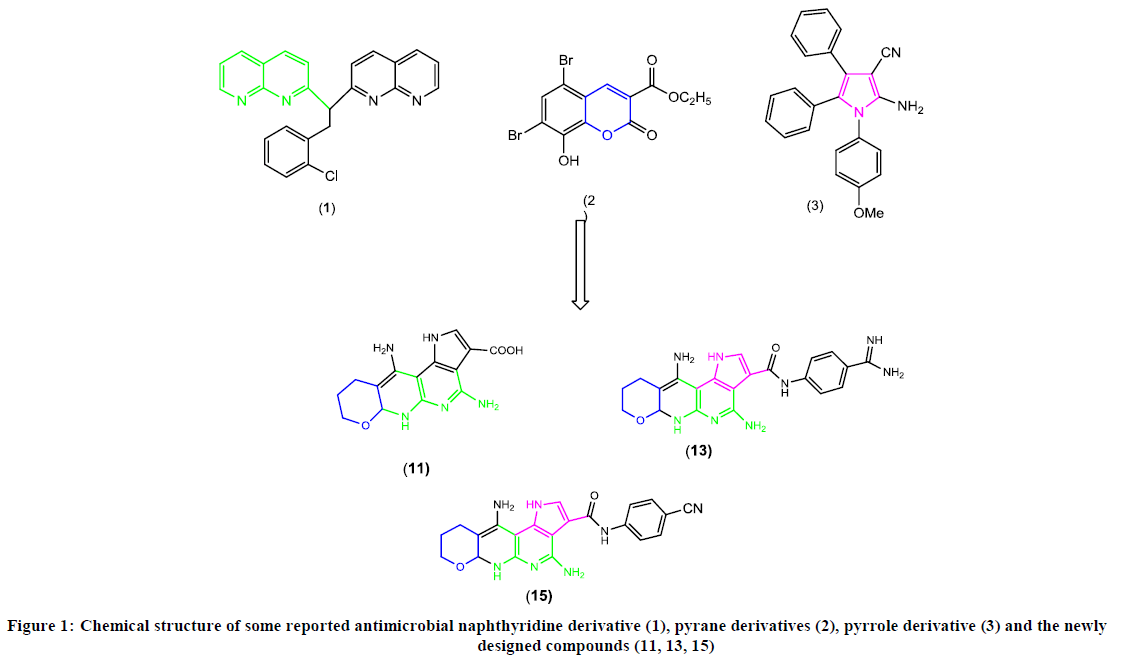
Figure 1: Chemical structure of some reported antimicrobial naphthyridine derivative (1), pyrane derivatives (2), pyrrole derivative (3) and the newly designed compounds (11, 13, 15)
Biologically active heterocyclic [21-27], we decided to hybridize these important scaffolds to get novel in the view of the aforementioned facts and in continuation of our work in the synthesis of compounds of expected antimicrobial activity (Figure 1). The present work has resulted in the formation of pyrano [2, 3-b] pyridine derivatives and pyrano pyrrolo naphthyridine derivatives of expected antimicrobial activity.
Materials and Methods
Chemistry
Reagents were purchased from Sigma Aldrich (BayouniTrading Co. Ltd., Saudi Arabia) and used without further purification. Reaction progress was monitored by thin-layer chromatograph yon silica gel pre-coated F254Merck plates (Darmstadt, Germany). Spots were visualized by ultraviolet irradiation. Melting points were determined on a Gallenkamp electro thermal meltingpoint apparatus (Weiss-Gallenkamp, Loughborough, UK) and are uncorrected. Infrared (IR) spectra were recorded as potassium bromide disks using Burker-Vector 22 Fourier transform infrared spectrophotometer (Billerica, MA). The NMR spectra were recorded with a Varian MercuryVXR-300 NMR spectrometer (Palo Alto, CA) at 300 and 75 MHz for Proton Nuclear Magnetic Resonance (1H-NMR) and Carbon-13 Nuclear Magnetic Resonance (13CNMR) spectra, respectively, is using DMSO-d6 as solvents. Mass spectra were recorded on a Hewlett Packard MS-5988 spectrometer (Palo Alto, CA) at 70 eV. Elemental analyses were carried out at the Micro-analytical Center of Cairo University, Giza, Egypt.
Ethyl 5, 7-diamino-3, 4, 8, 8a-tetrahydro-2H-pyrano [2, 3-b] pyridine-6-carboxylate (5)
A mixture of 3,4-dihydro-2H-pyran 4 (0.84 g, 0.01 mol), urea (0.6 g, 0.01 mol), and ethylcyano acetate (1.13 g, 0.01 mol) in absolute ethanol (20 ml) containing few drops of piperdine as a catalyst was heated under reflux for 8 h. The precipitate formed was filtered of, purified and recrystallization from methanol to afford the corresponding compound 5 and recrystallization from ethanol to afford pale yellow crystals in 67% yield; M.P. 240-242°C; IR (KBr): ν (cm-1), 3100-3400 (NH, NH2), 1740 (CO ester); 1H-NMR (δ, DMSO-d6): 1.28 (t, 3H, CH3), 1.98 (t, 2H, CH2), 2.5 (m, 4H, CH2), 3.57(s, 1H, NH), 4.47 (q, 2H, CH2), 4.49 (s, 1H, CH-NH2), 4.41 (t, 2H, CH2O), 5.56 (s, 4H, 2NH2); 13C-NMR (DMSOd6): 15.12 (CH3), 62.44 (CH2ester), 21.83 (CH2), 63.69 (CH2), 18.38 (CH2), 56.84 (C-NH2), 156.77, 83.46, 167.3 (C=O), 104.73 (C-CO), 59.88 (CH-NH2); MS (m/z, %): 239 (M+, 45). Anal. Calcd for C11H17N3O3 (239.27): C, 55.22; H, 7.16; N, 17.56%. Found: C, 55.25; H, 7.18; N, 17.59%.
5,8-Diamino-3,4,10,10a-tetrahydro-2H-pyrano[2,3-b][1,8]naphthyridin-6(9H)-one (6)
A mixture of compound 5 (2.39 g, 0.01 mol) and acetonitrile (15 ml) in presence of acetic acid (10 ml) was heated under reflux for 10 h. Then solvent was removed and the residue was extracted withethyl ether followed by purification through recrystallization from ethanol to afford compound 6 as green yellow crystals in 60% yield; M.P. 270-272°C; IR (KBr): ν (cm-1), 3100-3400 (NH2, NH), 1660 (CO Pyridine.); 1H-NMR (δ, DMSO-d6): 1.56 (t, 2H, CH2), 2.58 (m, 2H, CH2), 4.44 (t, 2H, CH2O), 4.49 (s, 1H, CH=C), 4.67 (s, 1H, CH), 4.79 (s, 1H, NH), 5.58 (s, 4H, 2NH2), 12.66 (s, 1H, NH); 13C-NMR (DMSO-d6): 23.1 (CH2), 24.92 (CH2), 68.68 (CH2O), 87.71 (CHO), 115.83 (CH), 82.72-196.46 (CH=CH-NH2), 182.91 (C=O); (MS (m/z, %): 234 (M+, 40). Anal. Calcd for: C11H14N4O2 (234.25): C, 56.40; H, 6.02; N, 23.92. Found: C, 56.42; H, 6.07; N, 23.96%.
6-(2-Phenylhydrazinyl)-3,4,10,10a-tetrahydro-2H-pyrano[2,3-b][1,8]naphthyridine-5,8-diamine (8)
A mixture of 6 (2.34 g, 0.01 mol) and phosphorus ox chloride (1.52 ml, 0.01 mol) was heated for a few minutes. After cooling, toluene was added to the reaction mixture followed by evaporation to give oily substance. The oily product was poured onto ice cold water and the medium became alkaline by the addition of aqueous 10% ammonium hydroxide. This alkaline mixture was extracted with ethyl acetate and the organic layer was washed with water, dried over magnesium sulfate, evaporated to give crude 7 (70%). A mixture of this crude 7 (2.52 g, 0.01 mol), phenyl hydrazine (1.08 g, 0.01 mol) in ethanol (30 ml) was heated under reflux for 18 h. The reaction mixture was concentrated under reduced pressure and the obtained product was purified by chromatography with 50% ethyl acetate/heptane, followed by recrystallizationfrom methanol. Afford reddish crystals in 55% yield; M.P. 240-242°C; IR (KBr): ν (cm-1): 3100-3400 (NH2, NH); 1H-NMR (δ, DMSO-d6): 1.54 (t, 2H, CH2), 2.35 (t, 2H, CH2), 3.64 (t, 2H, CH2O), 4.43 (s, 2H, 2NH), 4.69 (s, 1H, CHO), 5.42 (s, 1H, NH), 5.69 (s, 1H, CH), 7.08-8.09 (m, 9H, Aromatic proton, 2NH2); 13C-NMR (DMSO-d6): 22.3 (CH2), 24.84 (CH2), 68.49 (CH2O), 94.42 (CHO), 117.42 (CH), 84.58-139.44 (CH-NH2), 157.34 (CHNH), 148.95, 113.23, 129.22, 122.81 (Aromatic); (MS (m/z, %): 324 (M+, 30). Anal. Calcd for: C17H20N6O (324.38): C, 62.95; H, 6.21; N, 25.91%. Found C, 62.97; H, 6.25; N, 25.94%.
Ethyl 4,11-diamino -1,6,6a,8,9,10-hexahydropyrano[2,3-b]pyrrolo[2,3-f][1,8] naphthyridine -3-carboxylate (10)
A mixture of compound 8 (3.24 g, 0.01 mol) and ethyl propiolate (0.98 g, 0.01 mol) in methanol (10 ml) was heated by reflux for 18 h. The solvent was removed and the product was purified by chromatography on silica gel using 50% ethyl acetate/cyclohexane to produce 9 (50%). A mixture of the latter 9 (3.53 g, 0.01 mol) and dimethylformamide (25 ml) was refluxed for 24 h. After cooling, the reaction was poured on ice cold water and then extracted with ethyl acetate. The organic layer was driedover magnesium sulfate, evaporated, and theresidue purified by chromatography with 5% methanol/methylene chloride to give 10 as yellowcrystals in 75% yield; M.P.180-182°C; IR (KBr): ν (cm-1) 3100-3400 (NH2, NH), 1739 (C=O); 1H-NMR (δ, DMSO-d6): 1.41 (t, 3H, CH3), 2.01 (m, 2H, CH2), 2.05 (t, 2H, CH2), 3.44 (t, 2H, CH2O), 4.44 (s, 1H, NH), 4.48 (q, 2H, CH2), 4.71 (s, 1H, CHO), 5. 41 (s, 1H, NH-pyrrole ), 7.57 (s, 1H, CH-pyrrole), 8.62 (s, 4H, 2NH2); 13C-NMR (DMSO-d6):14.33 (CH3), 22.1 (CH2), 24.82 (CH2), 68.48 (CH2O), 94.41(CHO), 108.69(CH), 117.41 (CH), 84.58-156.33(C -NH2), 157.32(CHNH), 106.82-159.42 (C=CH), 148.93, 113.24, 129.26, 122.84 (Aromatic); (MS (m/z, %):329 (M+, 20). Anal. Calcd for: C16H19N5O3 (329.35): C, 58.35; H, 5.81; N, 21.26%. Found: C, 58.37; H, 5.84; N, 21.27%.
4,11-Diamino-1,6,6a,8,9,10-hexahydropyrano[2,3-b]pyrrolo[2,3-f][1,8]naphthyridine-3-carboxylic acid (11)
A mixture of 10 (3.29 g, 0.01 mol) and 10% sodium hydroxide in methanol (10 ml) with was stirred for 18 h at room temperature. After evaporating the solvent, the resulting residue was extracted with methylene chloride and washed with water. The aqueous layer was removed, acidified with 1 M Hydrochloric acid and extracted with methylene chloride. After evaporation, the resulting residue was purified through recrystallization from water to give 11 as Pale green crystals in 55% yield; M.P. 200-202°C; IR (KBr): ν (cm-1): 1720 (C=O), 3106-3402 (NH2, NH), 3450 (OH); 1H-NMR (δ, DMSO-d6): 2.01 (m, 2H, CH2), 2.05 (t, 2H, CH2), 3.44 (t, 2H, CH2O), 4.44 (s, 1H, NH), 4.71 (s, 1H, CHO), 5. 41 (s, 1H, NH-pyrrole ), 7.57 (s, 1H, CH-pyrrole), 8.62 (s, 4H, 2NH2), 11.47 (brs, 1H, OH); 13C-NMR (DMSO-d6), 61.47 (CH2), 22.9 (CH2), 24.89 (CH2), 68.48 (CH2O), 94.46 (CHO), 108.64 (CH), 117.48 (CH), 84.54-156.35(C -NH2), 157.39 (CHNH), 106.82-159.42 (C=CH), 148.93, 113.24, 129.26, 122.85 (Aromatic); (MS (m/z, %): 301 (M+, 16). Anal. Calcd for: C14H15N5O3 (301.30): C, 55.81; H, 5.02; N, 23.24%. Found: C, 55.84; H, 5.05; N, 23.26%.
4,11-Diamino-N-(4-carbamimidoylphenyl)-1,6,6a,8,9,10-hexahydropyrano[2,3-b]pyrrolo[2,3-f][1,8]naph-thyridine-3-carboxamide (13)
A solution of 4,11-diamino-1,6,6a,8,9,10-hexahydropyrano[2,3-b]pyrrolo[2,3-f][1,8] naphthyridine -3-carboxylic acid (11) (3.01 g, 0.01 mol), 4-aminobenzamidine dihydrochloride (12) (2.08 g, 0.01 mol), benzotriazol-1-yloxytris(pyrrolidino), phosphoniumhexa fluorophosphate (5.20 g, 0.01 mol) and N-ethyldiisopropylamine (1.29 g, 0.01 mol) in dimethylformamide (10 ml) was stirred at room temperaturefor 24 h. The mixture was poured on ice cold water followed by an extraction with ethyl acetate. The organic layer was removed and then treated with 1 M HCl acid. Separation and neutralization of the aqueous layer with sodium bicarbonate, followed by extraction with ethyl acetate gave a solution of 13 which was evaporated in vacuum afforded yellow crystals in 45% yield; M.P.: 225-227°C; IR (KBr): ν (cm-1): 1727(C=O), 3150-3410 (NH2, NH); 1H-NMR (δ, DMSO-d6): 2.32 (t, 2H, CH2), 2.62 (t, 2H, CH2), 3.45 (t, 2H, CH2O), 4.75 (s, 1H, CHO), 5.29 (s, 1H, NH), 5. 53 (s, 1H, NH-pyrrole ), 7.45 (s, 1H, CH-pyrrole), 7.01-8.02 (m, 4H, Aromatic protons), 8.45 (s, 2H, NH2), 9.26 (s, 4H, NH2), 9.43 (s, 2H, 2NH); 13C-NMR (DMSO-d6): 61.43(CH2) 22.12 (CH2), 24.85 (CH2), 68.44 (CH2O), 94.45 (CHO), 108.47 (CH), 117.47 (CH), 84.58-156.33 (C -NH2), 157.12 (CHNH), 106.87-159.32 (C=CH), 148.83, 113.24, 129.26, 122.84 (Aromatic); ( MS (m/z, %):418 (M+, 30). Anal. Calcd for: C21H22N8O2 (418.45): C, 60.28; H, 5.30; N, 26.78%. Found 60.30; H, 5.34; N, 26.82%.
4,11-Diamino-N-(4-cyanophenyl)-1,6,6a,8,9,10-hexahydropyrano[2,3-b]pyrrolo[2,3-f][1,8]naphthyridine-3-carboxamide(15)
A mixture of 4-cyanoaniline (14) (1.18 g, 0.01 mol), benzotriazol-1-yloxytris (pyrrolidino)phosphonium-hexafluorophosphate (5.20 g, 0.01 mole) and N-ethyldiisopropylamine (1.29 g, 0.01 mole) was added to compound 11 (3.01 g, 0.01 mole) in dimethylformamide (10 ml). The reaction was stirred for 24 h at room temperature. After which it was diluted with 20 ml of water and extracted with methylenechloride. The organic layer was dried over magnesium sulfate, evaporated and the resulting residue purifiedby chromatography (20% methanol/methylene chloride) to give 15. Recrystallization from ethanol to afforded yellow crystals in 45% yield; M.P. 225-227°C; IR (KBr): ν (cm-1) 1726 (C=O), 2213 (CN), 3172-3442 (NH2, NH); 1H-NMR (δ, DMSO-d6): 2.05 (m, 2H, CH2), 2.53 (t, 2H, CH2), 3. 52 (t, 2H, CH2O), 4.42 (s, 1H, CHO), 5.34 (s, 2H, 2NH), 5.46 (s, 1H, NH-pyrrole ), 7.59 (s, 1H, CH-pyrrole), 7.01-8.02 (m, 4H, Aromatic protons), 8.72 (s, 2H, NH2), 9.35 (s, 2H, NH2); 13C-NMR (DMSO-d6), 61.47 (CH2), 22.2 (CH2), 24.84 (CH2), 68.58 (CH2O), 94.44 (CHO), 108.79 (CH), 118.41 (CH), 84.58-156.36 (C -NH2), 117 (CN), 157.72 (CHNH), 106.83-159.72 (C=CH), 148.95, 113.74, 129.76, 123.84 (Aromatic); (MS (m/z, %):401 (M+, 20). Anal. Calcd for: C21H19N7O2 (401.42): C, 62.83; H, 4.77; N, 24.42%. Found 62.85; H, 4.80; N, 24.44%.
Antimicrobial activity
Antimicrobial activity of the tested samples was determined using a modified Kirby–Bauer disk diffusion method[28]. Briefly, 100 μl of the test bacteria/fungi were grown in 10 ml of fresh media until they reached a count of approximately 108 cells/ml for bacteria or 105 cells/mL for fungi. One hundred microliter of a microbial suspension was spread onto agar plates corresponding to the broth in which they were maintained. Isolated colonies of each organism that might be playing a pathogenic role should be selected from primary agar plates and tested for susceptibility by disk diffusion method [29]. Many media are available, but NCCL recommends Mueller–Hinton agar due to it result in good batch-to-batch reproducibility. Disk diffusion method for filamentous fungi tested using approved standard method (M38-A) developed for evaluating the susceptibility of filamentous fungi to antifungal agents. Plates inoculated with filamentous fungi as Aspergillus flavus at 25°C for 48 h; gram (+) bacteria as Staphylococcus aureus, Bacillus subtilis; gram (–) bacteria as Escherichia coli, Pseudomonas aeuroginosa they were incubated at 35-37°C for 24-48 h and yeast as Candida albicans incubated at 30°C for 24-48 h, and then the diameters of the inhibition zone were measured in millimeters. The standard disk of Amoxicillin (Antibacterial agent), Fluconazole (antifungal agent), served as positive control for antimicrobial activity but filter disks impregnated with 10 μl of solvent (distilled water, chloroform, and DMSO) were used as negative control. The agar used is Meuller-Hinton agar that's strictly tested for composition and pH scale. Further, the depth of the agar within the plate could be a issue to be thought of within the disk diffusion technique. This technique is well documented, and normal zones of inhibition are determined for susceptible and resistant values. Blank paper disks (Schueicher and Schuel, Spain) with a diameter of 8 mm were impregnated 10 μl of test concentration of the stock solutions. When a filter paper disk impregnated with a tested chemical is placed on agar, the chemical will diffuse from the disk into the agar. This diffusion will place the chemical in the agar only around the disk. The solubility of the chemical and its molecular size will determine the size of the area of chemical infiltration around the disk. If an organism is placed on the agar, it will not grow in the area around the disk if it is susceptible to the chemical. This space of no growth round the disk is understood as a “zone of inhibition” or “Clear zone. For the disk diffusion, the zone diameters were measured with slipping calipers of the National Committee for Clinical Laboratory Standards. Agar primarily based ways like E take a look at and disk diffusion are often smart alternatives as a result of they are simpler and faster than broth-based methods.
Molecular docking study
The crystal structures of glutamate bound at COX-2 isoform (Protein Data Bank; PDB: ID 1gdo). Docking was done using London dG force and sophistication of the results was performed using force field energy. Preparation of the synthesized compounds for docking was attained via their 3D structure built by Molecular Operating Environment (MOE, Version 2005.06, Chemical Computing Group Inc., QC, and Canada). Definite procedures were in use before docking which include: 3D protonation of the structures, running conformational analysis using systemic search, selecting the least energetic conformer and applying the same docking protocol used with ligands. Docking for the synthesized compounds was applied.
Conclusion
In this article application methodology to promote the synthesis of naphthyridine derivatives by the reaction of 4,11-diamino-1,6,6a,8,9,10-hexahydropyrano[2,3-b]pyrrolo[2,3-f][1,8]naphthyridine-3-carboxylic acid 11 with-aminobenz-amidine-dihydrochloride, benzotriazol-1-yloxytris (pyrrolidino) phos -phonium hexafluorophosphate and N-ethyldiisopropylamine afford 13 and or Benzotriazol-1-yloxytris (pyrrolidino)phosphonium hexafluorophosphate, 4-cyanoaniline and N-ethyldiisopropylaminein 15 purified good and satisfied yield. The structures of synthesized compounds were established by IR, 1H-NMR, 13C-NMR and mass spectra. Antimicrobial activity of these compounds 5-8, 10, 11, 13 and 15 was evaluated against B. subtilis, S. aureus, E. coli, P. aeruginosa bacteria, A. flavus and C. albicans. The results revealed that the candidate (6) was found to be the most active against all the tested microorganisms.
Acknowledgment
The financial support from Chemistry Department, Faculty of Science, Aswan University, Egypt, Chemistry Department, Faculty of Science, Zagazig University, Zagazig, Egypt, Faculty of Pharmacy, Beni Suef University and Jouf University, Kingdom of Saudi Arabia is gratefully acknowledged.
References
- H. Soliman, M. Yousif, M. Said, N. Hassan, M.M. Ali, H.M. Awad, F.M. Abdel-Megeid, Der Pharma Chemica, 2014, 6, 394-410.
- E. Pontiki, D. Hadjipavlou-Litina, K. Litinas, O. Nicolotti, A. Carotti, Eur. J. Med. Chem., 2011, 46, 191-200.
- S.K. Srivastava, A. Jha, S.K. Agarwal, R. Mukherjee, A.C. Burman, Anti Cancer Agents in Med. Chem. (Formerly Curr. Med. Chem.-Anti-Cancer Agents), 2007, 7, 685-709.
- L. Fu, X. Feng, J.J. Wang, Z. Xun, J.D. Hu, J.J. Zhang, Y.W. Zhao, Z.B. Huang, D.Q. Shi, ACS Combinat. Sci., 2014, 17, 24-31.
- V. Kumar, M. Jaggi, A.T. Singh, A. Madaan, V. Sanna, P. Singh, P.K. Sharma, R. Irchhaiya, A.C. Burman, Eur. J. Med. Chem., 2009, 44, 3356-3362.
- A. Trifilieff, D. Wyss, C. Walker, L. Mazzoni and R. Hersperger, J. Pharmacol. Experim. Therap., 2002, 301, 241-248.
- T. Aboul-Fadl, F.A. Bin-Jubair, O. Aboul-Wafa, Eur. J. Med. Chem., 2010, 45, 4578-4586.
- G. Lebaut, F. Fouchard, B. Kutscher, P. Emig, I. Fleischhauer, J. Schmidt, S. Szelenyi, N-benzylindole and benzopyrazole derivatives with anti-asthmatic, anti-allergic, anti-inflammatory and immune modulating effect,US5965582A, US Grant, 1999, 5, 965, 582.
- G. Grover, S.G. Kini, Eur. J. Med. Chem., 2006, 41, 256-262.
- J. Matsumoto, T. Miyamoto, A. Minamida, Y. Nishimura, H. Egawa, H. Nishimura, J. Med. Chemi., 1984, 27, 292-301.
- N. Aggarwal, R. Kumar, P. Dureja, J.M. Khurana, Chem. Boil. Drug Design, 2012, 79, 384-397.
- K. Mogilaiah, G.R. Sudhakar, Synthe. Commun., 2003, 33, 3131-3134.
- A. Gopalsamy, K. Lim, G. Ciszewski, K. Park, J.W. Ellingboe, J. Bloom, S. Insaf, J. Upeslacis, T.S. Mansour, G. Krishnamurthy, J. Med. Chem., 2004, 47, 6603-6608.
- J.B. Shaik, B.K. Palaka, M. Penumala, S. Eadlapalli, M. Darla Mark, D.R. Ampasala, R. Vadde, D. Amooru Gangaiah, Chem. Boil. Drug Design, 2016, 88, 43-53.
- M.A. Mottaleb, S. Usenko, J.G. O’Donnell, A.J. Ramirez, B.W. Brooks, C.K. Chambliss, J. Chromatogra. A, 2009, 1216, 815-823.
- C.B. Sangani, D.C. Mungra, M.P. Patel, R.G. Patel, Chinese Chem. Lett., 2012, 23, 57-60.
- M.S. Al-Saleem, E.H. El-Sayed, M.A. El-Moneim, A. RE, Mens. Agitat., 2018, 13, 11-17.
- N.B. Dyatkina, C.D. Roberts, J.D. Keicher, Y. Dai, J.P. Nadherny, W. Zhang, U. Schmitz, A. Kongpachith, K. Fung, A.A. Novikov, J. Med. Chem., 2002, 45, 805-817.
- R.A. Rane, S.D. Gutte, N.U. Sahu, Bioorg. Med. Chem. Lett., 2012, 22, 6429-6432.
- M. Mohamed, R. El-Domany, R.A. El-Hameed, Acta Pharmaceutica., 2009, 59, 145-158.
- A.A. Ghoneim, N.A.A. Elkanzi, R.B. Bakr, Journal of Taibah University for Science, 2018, 12(6), 774-782.
- K.R. Abdellatif, R.B. Bakr, Bioorg. Chem., 2018, 78, 341-357.
- M.A. Abdelgawad, R.B. Bakr, A.O. El-Gendy, G.M. Kamel, A.A. Azouz, S.N.A. Bukhari, Fut. Med. Chem., 2017, 9, 1899-1912.
- K.R.A. Abdellatif, E.K.A. Abdelall, R.B. Bakr, Curr. Topics in Med. Chem., 2017, 17, 941-955.
- I.H. El Azab, N.A.A. Elkanzi, Synthe. Commun., 2014, 44, 2692-2714.
- N.A.A. Elkanzi, Asian J. Chem., 2014, 26(20), 6874-6878.
- A. Teplyakov, G. Obmolova, M.A. Badet-Denisot, B. Badet, Protein Sci., 1999, 8, 596-602.
- S.A. Komykhov, K.S. Ostras, A.R. Kostanyan, S.M. Desenko, V.D. Orlov, H. Meier, J. Heterocycl. Chem., 2005, 42, 1111-1116.
- M.A. Pfaller, L. Burmeister, M. Bartlett, M. Rinaldi, J. Clin. Microbial., 1988, 26, 1437-1441.
- N.A.A. Elkanzi, A.A. Ghoneim, R.B. Bakr, J. Heterocyclic Chem., 2019, 56, 406-416.



