Research Article - Der Pharma Chemica ( 2017) Volume 9, Issue 2
Design of New Inhibitors of Dipeptidyl Peptidase-4 in Type 2 Diabetes by Computer Simulations
Hicham Ayachi1, Shirin Jamshidi2, Meriem Merad1, Yassine Bendiabdallah3, Said Ghalem1 and Khondaker Miraz Rahman22Institute of Pharmaceutical Science, King’s College London, London, SE1 9NH, UK
3Laboratory (Dr Bendiabdallah), 53 Beauchamp Place, London SW3 1NY, UK
Abstract
The techniques of molecular modelling are widely used in chemistry, biology and the pharmaceutical industry. Most current drugs target enzymes. This theoretical approach allows us to predict the mode of interaction of a ligand with its receptor. Inhibition of Dipeptidyl Peptidase-4 “DPP-4” is an important approach in the treatment of disease in Type 2 diabetes. Several inhibitors have already been identified, but their affinity is insufficient to consider any of them being a pharmacological development. High-affinity inhibitors are used to inhibit DPP-4 as Linagliptin, Sitagliptin, Vildagliptin, Saxagliptin and Alogliptin but also as an adjuvant therapy for the treatment of Type 2 diabetes. It is for this purpose that molecular modelling techniques like docking and molecular dynamics have been developed. The results obtained in this work show the inhibition of DPP-4 by molecular modelling methods. The introduction of bulky groups causes a conformational rearrangement in the pocket of the active site that will probably be strengthened by the complementarity and therefore the activity increases. The results obtained in this study, by methods of molecular modelling which have been elucidated, allowed us to conclude that Linagliptin is a better inhibitor of DPP-4 than Sitagliptin, Vildagliptin, Saxagliptin and Alogliptin. Linagliptin has the potential to be the best inhibitor of DPP-4 in the treatment of Type 2 diabetes based on molecular modeling interaction.
Keywords
DPP-4, Type 2 diabetes, Inhibitor, Molecular docking, Molecular dynamics simulations
Introduction
Diabetes is a metabolic disease that poses a major problem to public health, with it causing nearly four million deaths each year worldwide [1]. Type 2 diabetes is a condition of the general metabolism of carbohydrates, fats and proteins characterized by an abnormal increase in sugar levels in the blood (hyperglycemia). This condition is due to defective insulin secretion, itsinaction, or both combined [2]. Hyperglycemia is most often associated to more or less evocative external symptoms of disease severity. In addition to acute complications (hyperglycemia, ketoacidosis, hyperosmolar syndrome), hyperglycemia degenerates on more or less severe degenerative complications [3,4]. New approaches coming in full light are, for some years, on the market. They target Glucagon-like peptide-1 “GLP1”. It is a digestive hormone helping the body to normalize blood glucose levels when it rises abnormally. However, the half-life of this hormone is very short due to its degradation by an enzyme: DPP-4 thus making its potential as a therapeutic agent considerably less. To work around this obstacle, two strategies have been adopted: firstly, the injectable exogenous development of analogues of GLP- 1 resistant to the action of DPP-4, and secondly the use of oral medications that selectively block DPP-4 to extend the endogenous GLP-1 half-life [5,6]. We are interested in the latter, newer approach, aiming to treat Type 2 Diabetes by inhibiting DPP-4. DPP-4 is a relatively small molecule: therefore selectively inhibiting DPP-4 contributes significantly to normalize blood glucose levels with very few side effects [7].
Materials and Methods
Dipeptidyl peptidase-4 (DPP-4) protein
DPP-4 consists of two broken intestinal hormones called incretins. The incretins are produced in the gut when food is consumed, and they stimulate insulin secretion, which lowers glucose levels in the blood. DPP-4 is an N-terminal dipeptidyl exopeptidase which comes as both a membrane-bound protein and as a soluble protein in plasma. Low-molecular weight inhibitors of DPP-4 have been investigated for many years. DPP-4 inhibitors lower blood glucose, improve glucose tolerance and ameliorate insulin response to glucose associated with patients in type 2 diabetes. Medicines to treat Type 2 diabetes have been developed that inhibit DPP-4, preventing the degradation of incretin and prolong insulin secretion, increasing its effect (Figure 1) [8].
DPP-4 was downloaded from the database Bookhaven Protein Data Bank (access code 3F8S). The three dimensional structure of Dipeptidyl peptidase-4 was obtained by X-ray diffraction with a resolution (2.43 Å) [9].
Inhibitors of DPP-4
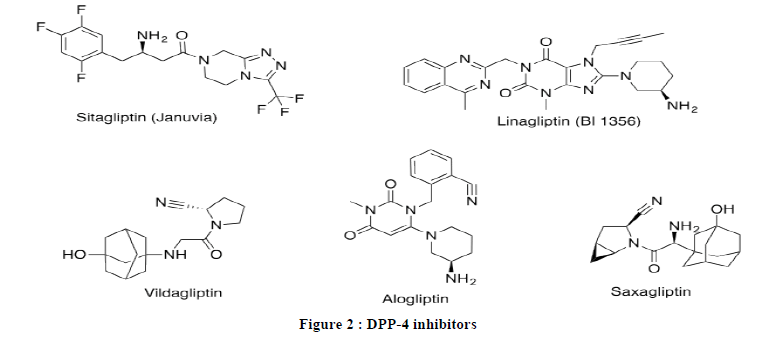
The following DPP-4 inhibitors Linagliptin, Sitagliptin, Vildagliptin, Saxagliptin, Alogliptin that are used to treat type 2 diabetes were selected for the study which were previously compared in a similar study (Figure 2) [10].
Methods
Treatment of protein
The protein to be used should be first processed using the following steps:
- Downloading proteins from the database Bookhaven Protein Data Bank (3F8S) [9].
- Removeng the water molecules to stabilise the protein.
- Eliminating Co-crystallisation of molecules to achieve a simplified model.
- Identifying the active site of the protein.
Construction of ligands
The ligands used in this work are drawn with Hyperchem8 software. A step of optimising the geometry becomes necessary. For this, we applied the semi-empirical method (AM1) [11]. The molecules thus obtained are recorded with pdb or mol.
Molecular docking and complex construction
The next step after the construction of ligands, and the positioning of these molecules in the active site of the enzyme (3F8S). For this, we used the GOLD [12] software, the ligand-receptor complex is formed, it will adapt to the most stable conformation, i.e., the one with the lowest energy level.
Conclusion
The results obtained in this study, by the methods of molecular modeling were elucidated, allowing us to conclude that Linagliptin is a better inhibitor of DPP-4. Linagliptin inhibits DPP-4 more effectively than Sitagliptin, Vildagliptin, Saxagliptin and Alogliptin. Linagliptin has the potential to be the best inhibitor of DPP-4 in the treatment of type 2 diabetes.
References
[1] A. Slama-Chaudhry, M. Mavromati, A. Golay, 2013, DMCPRU, P29,
[2] F. Couic-Marinier, 2009, 48, 34-37.
[3] A. Boudiba, S. Mimouni-Zerguini, 2008, 53, 19-21.
[4] S.M. Haffiner, M.P. Stern, H.P. Hazuda, B.D. Mitchell, J.K. Patterson. J. Am. Med. Assoc., 1990, 263, 2893-2898.
[5] S. Halimi, I. Debaty, L. Villaret, M. Muller. La. Rev. De. Méd. Int., 2008, 29, 881-890.
[6] J.F. Gautier, S.P. Choukem. Nutrition Clinique et Métabolisme., 2008, 22, 59-65.
[7] R. Kurukulasuriya, J.J. Rohde, B.G.Szczepankiewicz, F. Basha, C. Lai, H.S. Jae, Bioorg. Med. Chem. Lett., 2006, 16, 6226-6230.
[8] F.P. Miller, A.F. Vandome, J.M. Brewster, 2010, P80.
[9] R.G. Ewan, Y. Xiong, J. Melanie, L. D’andrea, L. Regan, Elsevier Science Ltd., 2003, 11, 497-508.
[10] L. Thomas, M. Eckhardt, E. Langkopf, M. Tadayyon, F. Himmelsbach, M. Mark, J. Pharmacol. Exp Therapeut., 2008, 325, 175-182.
[11] M.J.S. Dewar, E.G. Zoebisch, E.F. Healy, J.J.P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902-3909.
[12] G. Jones, P. Willett, R.C. Glen, A.R. Leach, R. Taylor. J. Mol. Biol., 1997, 267, 727-748.
[13] P. Labute, C. Williams, M. Feher, E. Sourial, J.M. Schmidt, J. Med. Chem., 2001, 44, P1483-1490.
[14] http://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/
[15] https://sourceforge.net/projects/smina/files/
[16] W. Soufi, M. Merad, F. Boukli, S. Ghalem, Adv. Mol. Imaging., 2011, P17-P23.
[17] S. Teniou, 2012, 57-67.



