Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 3
DFT-QSAR and Docking Studies of 2-[5-(aryloxymethyl)-1,3,4-oxadiazol-2-ylsulfanyl] acetic acids Derivatives against Bacillus subtilis
Abel K. Oyebamiji* and Semire Banjo
Department of Pure and Applied Chemistry, Faculty of Pure and Applied Sciences, Ladoke Akintola, University of Technology, P.M.B-4000, Ogbomoso, Oyo State, Nigeria
- *Corresponding Author:
- Abel K. Oyebamiji
Department of Pure and Applied Chemistry
Faculty of Pure and Applied Sciences
Ladoke Akintola, University of Technology
P.M.B-4000, Ogbomoso, Oyo State, Nigeria
Abstract
Density Functional theory (DFT), Quantitative Structure Activity Relation (QSAR) and docking methods were used to observed the anti-bacteria activity of 2-[5-(aryloxymethyl)-1,3,4-oxadiazol-2-ylsulfanyl] acetic acids derivatives. Several descriptors such as Highest Energy Occupied Molecular Orbital (HOMO), Lowest Unoccupied Molecular Orbital (LUMO), LogP, molecular weight, dipole moment, chemical hardness, chemical potential and solvation energy obtained via DFT method were employed to develop QSAR model. This was used to predict the bioactivity (IC50) that fitted well with the observed IC50. More so, every studied compound was docked against B. subtilis cell line (1a6f) and the binding energy obtained from ligand-receptor interactions were reported.
Keywords
2-[5-(aryloxymethyl)-1,3,4-oxadiazol-2-ylsulfanyl] acetic acids derivatives, DFT-QSAR, Molecular docking
Introduction
Bacteria are most numerous organism formed in the universe. They are commonly found amidst man and they are advantageous to man [1-3]. They are very trivial to see with human eye unless it is assisted; this is because they belong to the class of prokaryotic cellular organization. They are numerous in human body than the normal human-cells; thus, they are crucial for apposite enhancement, sustenance and resistance to illness [4,5].
Many years ago, Bacillus subtilis, a constricted group of closely related sorts has been acknowledged not able to be distinguished on the root of phenotypic physiognomies [6]. This genus called “Bacillus” comprised rod-shaped bacteria as well as bacteria that form endospore [7,8]. Bacillus is a Gram-positive species that is heterogeneous in nature and it is also a facultative anaerobic bacteria which can be controlled by bacteriophages because of their highest occurrence in food production [9,10]. According to Pedersen et al., it is observed that B. subtilis produces enterotoxins and emetic Toxin which lead to food-borne diseases [11]. More so, B. subtilis is more frequently met in the surroundings [12].
Moreover, several years ago, acetic acid was first isolated through distillation of vinegar [13]. Due to its distinctive whiff and acerbic taste of vinegar, it shows various features in its pure state i.e., Colourless with a severe and maddening scent of vinegar [14-16]. Most of the derivatives of acetic acid proves to be useful in the manufacturing of chemicals, antibiotics, plastics, synthetic fibers and explosives [17,18].
Furthermore, Quantitative Structural Activity Relationship (QSAR) which is an arithmetic model helps to predict the toxicity of substance and also render support in the case of classic chemicals [19-21]. In this QSAR model, molecular descriptors which described anti-bacteria activity of 2-[5-(aryloxymethyl)-1,3,4-oxadiazol-2-ylsulfanyl] acetic acids Derivatives such as Highest Energy Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) energy, band gap energy, dipole moment, charges on every heteroatoms etc. [22-24] were obtained by using Quantum Chemical Methods (QCM) via Density Functional Theory (DFT). This has been described to be suitable for creating sufficient QSAR model. Thus, quantum chemical descriptors usage has prodigious significance [25-28].
In the past decade, docking has a means of ascertaining the interaction between ligand and receptor have received a great number of attention. This is due to its ability to practically select enormous set of compounds and scoring as well as exposing the stages involved in prevention of target binding site by ligands [29-31]. Thus, interaction energy calculations can be presented dock score form [32].
Therefore, as shown in Figure 1, Neelam et al., synthesized and screened seven compounds against B. subtilis and then optimized by using density functional theory method [33]. These studied compounds are: 2-(5-(phenoxymethyl)-1,3,4-oxadiazol-2-ylsulfanyl) acetic acid (3a), 2-(5- ((4-methylphenoxy) methyl)-1, 3, 4-oxadiazol-2-ylsulfanyl) acetic acid (3b), 2-(5-((4-methoxyphenoxy) methyl)-1, 3, 4-oxadiazol-2-ylsulfanyl) acetic acid (3c), 2-(5-((4-chlorophenoxy)methyl)-1,3,4-oxadiazol-2-ylsulfanyl) acetic acid (3d), 2-(5-((4-bromophenoxy)methyl)-1,3,4-oxadiazol-2-ylsulfanyl) acetic acid (3e), 2-(5-((4-fluorophenoxy)methyl)-1,3,4-oxadiazol-2-ylsulfanyl) acetic acid (3f) and 2-(5-((4- nitrophenoxy)methyl)-1,3,4-oxadiazol-2-ylsulfanyl) acetic acid (3g). So, the aim of this work is to calculate molecular parameters by using DFT method and then build QSAR model for investigation of bioactivity of chosen molecules. Therefore, the optimized compound were docked together with receptor (1a6f) so as to obtain free binding energy.
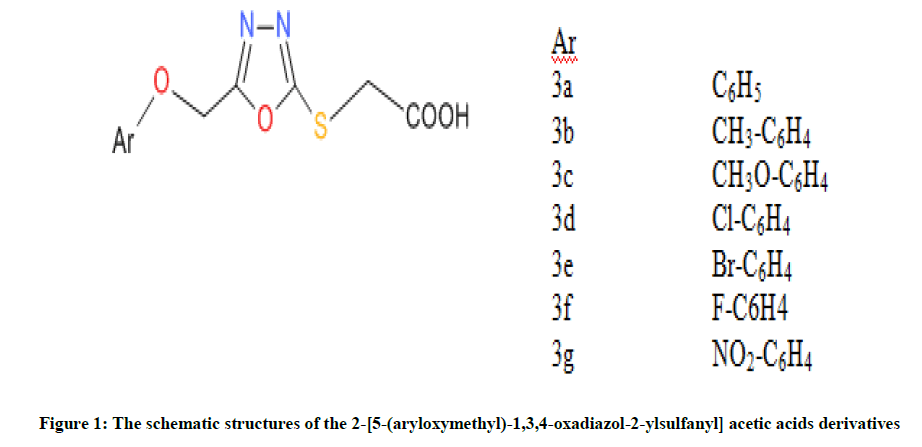
Figure 1: The schematic structures of the 2-[5-(aryloxymethyl)-1,3,4-oxadiazol-2-ylsulfanyl] acetic acids derivatives
Computational details
Molecular descriptors and QSAR studies
The optimization of equilibrium geometries for seven molecular compounds was executed via quantum chemical method. In this paper, optimization of the molecular compounds was carried out by using 6-31(d,p) basis set for the calculation of molecular descriptors which describe the bioactivity of the compounds. This was achieved using quantum chemical software i.e. Spartan ’14 by wave function Inc (Spartan 14) [34].
Moreover, the obtained molecular descriptors were used to build QSAR model for biological investigation of the selected molecules [35]. Multiple linear regression method, a frequent arithmetic method was used in building the QSAR model and was then validated by considering cross validation (R2) and adjusted R2 (Equations 1 and 2):
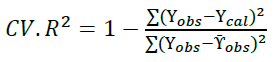 (1)
(1)
The R2 adjusted could be calculated using Equation 2:
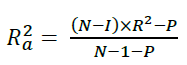 (2)
(2)
Moreover, the optimized molecular structures were also used for docking study to estimate binding affinity of the molecules to the B. subtilis cell line, MTCC 96 receptor (PDB ID: 1a6f). The docking software used to achieve this were Discovery Studio, Autodock tool, AutoDock vina programme and Edu pymol version 1.7.4.4 which was used as post dock software. More so, the grid dimension used for 1a6f was 32 × 32 × 36 Å (grid size) together with 1.000 Å as the grid-point spacing.
Result and Discussion
QSAR modeling
In this study, both dependent and independent variables were involved in the development of QSAR model via Multiple Linear Regression (MLR) method. More so, the obtained molecular parameters served as the independent variable while Inhibitory Concentration (IC50) against B. subtilis cells line as dependent variable. The selected descriptors as shown in Table 1 were used to build the QSAR model, since the quality of any developed QSAR model is a function of it predictability power. Therefore, it is noted that the developed model replicated the experimental IC50 well as revealed by the fitting factor (R2) (Figure 2). The calculated fitting factor (R2), 0.9996, revealed the adeptness of the model as shown in Equation 3.
| HOMO | LUMO | BG | DM (Debye) | SE (Kj/mol) | CH | CP | GN | LOGP | MW | OVALITY | PSA | HET | POL | HBA | HBD | B. subtilis (MTCC 96) |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3a | -6.46 | -0.85 | 5.61 | 4.54 | -48.49 | 2.80 | -3.66 | 2.38 | 3.20 | 252.25 | 1.41 | 68.15 | -1.054 | 58.09 | 6 | 1 | 0.60 |
| 3b | -6.30 | -0.82 | 5.48 | 4.67 | -47.41 | 2.74 | -3.56 | 2.31 | 3.69 | 266.27 | 1.44 | 68.19 | -1.054 | 59.60 | 6 | 1 | 0.55 |
| 3c | -5.94 | -0.78 | 5.16 | 3.90 | -52.66 | 2.58 | -3.36 | 2.19 | 3.07 | 282.27 | 1.46 | 75.14 | -1.058 | 60.40 | 7 | 1 | 0.55 |
| 3d | -6.52 | -0.99 | 5.53 | 4.71 | -46.76 | 2.77 | -3.76 | 2.55 | 3.76 | 286.69 | 1.44 | 68.00 | -1.059 | 59.22 | 6 | 1 | 0.50 |
| 3e | -6.52 | -1.03 | 5.49 | 4.65 | -48.21 | 2.75 | -3.76 | 2.57 | 4.03 | 331.14 | 1.44 | 68.07 | -1.055 | 59.59 | 6 | 1 | 0.50 |
| 3f | -6.44 | -0.9 | 5.54 | 4.57 | -42.20 | 2.77 | -3.67 | 2.43 | 3.36 | 270.24 | 1.42 | 68.04 | -1.061 | 58.48 | 6 | 1 | 0.45 |
| 3g | -6.14 | -0.89 | 5.25 | 4.68 | -55.09 | 2.62 | -3.52 | 2.35 | 2.84 | 299.26 | 1.48 | 116.63 | -1.056 | 60.43 | 9 | 3 | 0.40 |
Table 1: The calculated molecular descriptors obtained from the studied compounds
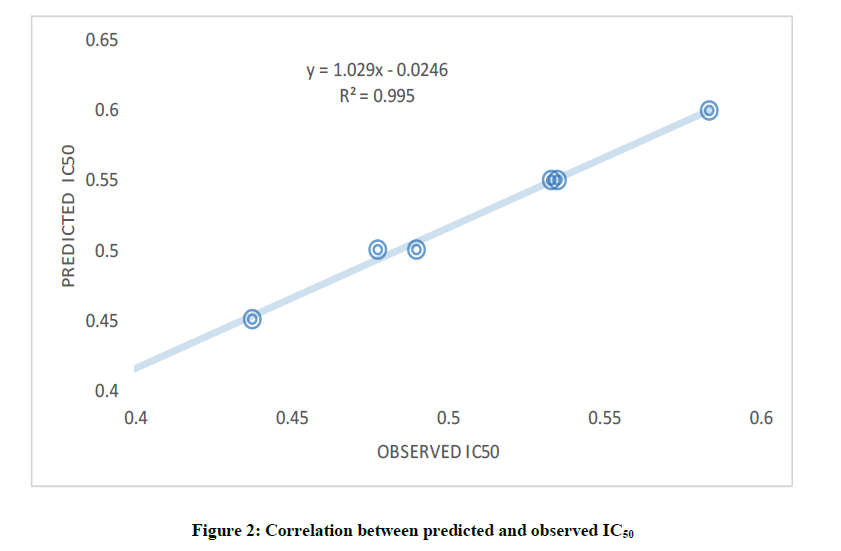
Figure 2: Correlation between predicted and observed IC50
Furthermore, Cross Validation (CV.R2) and adjusted R2 were considered in order to statistically validate the developed model. The CV.R2 was calculated to be 0.915 which was observed to be greater than the standard value (0.5) [36] and this disclosed the dependability and appropriateness of the developed model as well as the adjusted R2 (0.990) which was greater than 0.6 (Table 2).
| N | p | R2 | CV.R2 | R2adj |
|---|---|---|---|---|
| 7 | 4 | 0.996 | 0.915 | 0.990 |
Table 2: Statistical parameters for validation of QSAR model
IC50 = -1.648 – 0.017(SE) – 0.092(HBD) – 0.458(HOMO) – 0.613(GN) (3)
The predicted anti-bacteria activity of the ligands using the QSAR model as well as deviation from the experimental values were displayed in Table 3.
| Compounds | Observed | Predicted | Residual |
|---|---|---|---|
| 3a | 0.6 | 0.6 | 0 |
| 3b | 0.55 | 0.55 | 0 |
| 3c | 0.55 | 0.55 | 0 |
| 3d | 0.5 | 0.49 | 0.01 |
| 3e | 0.5 | 0.51 | -0.01 |
| 3f | 0.45 | 0.45 | 0 |
| 3g | 0.4 | 0.4 | 0 |
Table 3: Stepwise regression result for anti-bacteria activity
Docking and scoring
Investigation was carried out on the interaction between 2-[5-(aryloxymethyl)-1,3,4-oxadiazol-2-ylsulfanyl] acetic acids derivatives and B. subtilis cell line (1a6f) [37] which was obtained from protein data bank and from this investigation, nine conformations were observed. From this docking study, highest free binding energy (i.e., More negative value) in every docking simulation which was presumed to be most stable were reported. Therefore, the free binding energy calculated for compound 3a-3g are -5.4 kcal/mol, -5.8 kcal/mol, -5.6 kcal/mol, -5.6 kcal/mol, - 5.7 kcal/mol, -5.6 kcal/mol and -5.7 kcal/mol (Table 4).
| Comp. | Affinity (kcal/mol) |
H-Bond Between protein residues in the binding pocket and Drug | Distance |
|---|---|---|---|
| 3a | -5.4 | (i) LYS-11, LIG: O (ii) LEU-48, LIG:O (iii) VAL-46, LIG:O (iv) LEU-10, LIG:H | (i) 3.4 (ii) 2.4 (iii) 2.8 (iv) 2.8 |
| 3b | -5.8 | (i)ARG-68, LIG: O (ii) LEU-10, LIG:O (iii) VAL-46, LIG:O | (i)2.2, (ii) 2.9, (iii) 2.7 |
| 3c | -5.6 | (i) ASN-13, LIG: O (ii) LYS-11, LIG:N (iii) LEU-10, LIG:O (iv) VAL-46, LIG:H (v) ARG-68 LIG:O | (i) 2.2, (ii) 3.2, (iii) 2.9, (iv) 2.7, (v) 2.3 |
| 3d | -5.6 | (i) LYS-11, LIG:N (ii) LEU-48, LIG:O (iii) LEU-10, LIG:O (iv) VAL-46, LIG:O (v) LEU-48, LIG: O | (i) 3.2, (ii) 3.3, (iii) 2.8, (iv) 2.8, (v) 2.7 |
| 3e | -5.7 | (i) ALA-2, LIG:O (ii) ARG-65, LIG:O (iii) HIS-3, LIG:O (iv) ARG-9, LIG: O (v) ARG-68, LIG:N (vi) ARG-9, LIG:O | (i) 2.7, (ii) 2.0, (iii) 3.3, (iv) 1.8, (v) 2.1, (vi) 2.4 |
| 3f | -5.6 | (i) LYS-11, LIG:O (ii) LEU-10, LIG:O (iii) VAL-46, LIG:O (iv) LEU-48, LIG:O | (i) 3.4, (ii) 2.7, (iii) 2.7, (iv) 2.7 |
| 3g | -5.7 | (i) ASN-13, LIG: H (ii) LYS-11, LIG:O (iii) LYS-11, LIG:O (iv) LEU-10, LIG: O (v) VAL-46, LIG: O (vi) ARG-68, LIG: O | (i) 2.5, (ii) 3.4, (iii) 3.4, (iv) 3.0, (v) 2.8, (vi) 2.4 |
Table 4: Interactions between ligands and 1a6f receptor
Moreover, as shown in Figure 3, exploration of the ligand-receptor complex revealed that H-bonds interaction played a protuberant role in the binding of ligand to the enzyme which then affect it biological functions. So, Table 3, show the H-bonds and the distances in the ligand-receptor complex. In this study, the residues involved in the interaction for each compound are; residue LYS-11, LEU-48 and VAL-46 formed hydrogen bonds with compound 3a; for 3b, ARG-68, LEU-10 and VAL-46 were involved in the interaction. Moreover, the residues that took part in hydrogen bond interaction for compound 3c, 3d, 3e, 3f and 3g are ASN-13, LYS-11, LEU-10, VAL-46, ARG-68; LYS-11, LEU-48, LEU-10, VAL-46, LEU-48; ALA-2, ARG-65, HIS-3, ARG-9, ARG-68, ARG-9; LYS-11, LEU-10, VAL-46, LEU-48; ASN-13, LYS-11, LYS-11, LEU- 10, VAL-46, ARG-68 respectively. The selected ligand-receptor (1a6f) complexes showing stable conformation were displayed in Figure 3. Thus, it was presumed that compound 3b inhibited 1a6f more than all other compounds and this could be a function of the structure of compound 3b with the effect of Methylbenzene as the derivative as shown in Figure 1.
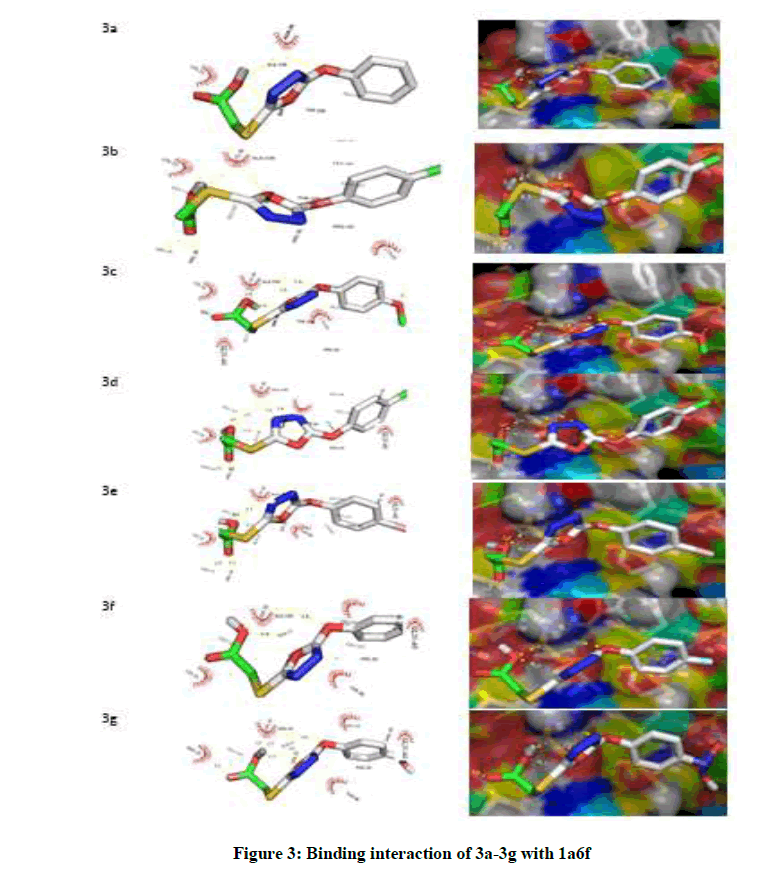
Figure 3: Binding interaction of 3a-3g with 1a6f
Conclusion
In this paper, anti B. subtilis activity of 2-[5-(aryloxymethyl)-1,3,4-oxadiazol-2-ylsulfanyl] acetic acids derivatives was achieved by using the obtained descriptors with the use of density functional theory method for QSAR study. The developed QSAR model reproduced the experimental bioactivities. More so, the results gotten from docking study predicted stable conformations of the ligands within the enzyme’s active gouge.
References
- F. Backhed, R. Ley, J. Sonnenburg, D. Peterson, J. Gordon, Science., 2005, 307, 1915-1920.
- A.S. Fauci, E.A. Zerhouni, Science., 2005, 308, 49.
- J. Kaiser, Science., 2005, 308, 938.
- S. Maloy, M. Schaechter, Int. Microb., 2006, 9, 1-7.
- K. Overbye, J. Barrett, Drug Discovery Today., 2005, 10, 45-52.
- P.R. Alejandro, P.J.P. Neil, E. Christopher, L.S. James, D.B. Jason, Int. J. Syst. Evol. Microbiol., 2009, 59, 2429-2436.
- C.T. Hou, D.P. Labeda, A.P. Rooney, Antonie Van Leeuwenhoek., 2005, 88, 167-171.
- N.P.J. Price, A.P. Rooney, J.L. Swezey, E. Perry, F.M. Cohan, FEMS Microbiol Lett., 2007, 271, 83-89.
- H. Shin, N. Bandara, E. Shin, S. Ryu, K.P. Kim, Research in Microbiol., 2011, 162(8), 791-797.
- T. Nagai, F. Yamasaki, Food Sci. Technol. Res., 2009, 15(3), 293-298.
- P.B. Pedersen, M.E. Bjornvad, M.D. Rasmussen, J.N. Petersen, Regul. Toxicol. Pharmacol., 2002, 36(2), 155-161.
- F. Cohn, Post Biol Plant., 1872, 1, 127-244.
- C. Sallmelin, J. Hovinen, J. Vilpo, Mutat. Res. Genet. Toxicol. Environ. Mutagen., 2000, 467, 129-138.
- K. Hiraoka, R.Y. Yamdagni, P, Kebarle, J. Am. Chem. Soc., 1973, 95, 6834.
- A. Kusakiewcz-Dawid, M. Bugaj, J.M. Dzik, B. Golos, P. Winska, K. Pawelczak, B. Rzeszotarska, W. Rode, Acta Biochim. Pol., 2002, 49, 197-203.
- A. Fukumizu, Y. Tsuda, K. Wanka, M. Tada, S. Okamoto, A. Hijikata-Okunomiya, Y. Okada, Chem. Pharm. Bull., 1999, 47, 1141-1144.
- C. Tample, A. Stewartt, D. Meredith, N. Lister, K. Morgan, I. Collier, R. Vaughan-Jones, C.A.R. Boyed, P. Bailey, J.R. Brank, Communication., 1998, 273, 20-22.
- A.W. Bauer, M. M. Kirby, J.C. Sherris, M. Turck, Am. J. Clin. Pathol., 1996, 45, 493-496.
- G.E. Dahl, N. Jaitly, R. Salakhutdinov, arXiv preprint arXiv., 2014, 1231.
- A.K. Oyebamiji, B. Semire, Cancer Biol., 2016, 6(2), 69-72.
- K.A. Oyebamiji, B. Semire, New York Sci. J., 2016, 9(6).
- C. Hansch, Acc. Chem. Res., 1969, 2, 232-239.
- C.A. Ramsden, Quantitative Drug Design of comprehensive Medicinal Chemistry., 1990, 4.
- A.K. Oyebamiji, B. Semire, Bull. Pharm. Res., 2016, 6(3), 105-13.
- S. Arulmozhiraja, M. Morita, Chem. Res. Toxicol., 2004, 17, 348-356.
- C.G. Gu, X. Jiang, X.H. Ju, X.L. Yang, Y.R. Bian, C. Sun, Ecotoxicol Environ Saf., 2009, 72, 60-70.
- E. Eroglu, H. Turkmen, J. Mol. Graph Model., 2007, 26, 701-708.
- M. Zhu, F. Ge, R. Zhu, X. Wang, X. Zheng, Chemosphere., 2010, 80, 46-52.
- J. Cherfils, J. Janin, Curr. Opin. Struct. Biol., 1993, 3, 265-269.
- I.D. Kuntz, E.C. Meng, B.K., Shoichet, Acc. Chem. Res., 1994, 27, 117-123.
- V. Sharma, S.R. Wakode, V. Lather, R. Mathur, M.X. Fernandes, Bull Pharm. Res., 2011, 1(2), 33-40.
- G.M. Morris, M. Lim-Wilby, Mol. Biol., 2008, 443, 365-382.
- J. Neelam, S. Deepika, G. Ruchika, J. Sandeep, Der Pharma Chemica., 2016, 8(4), 434-438.
- Spartan 14 wave function Inc. Irvine, CA 92612, USA
- E. Pourbasheer, S. Riahi, M.R. Ganjali, P. Norouzi, Eur. J. Med. Chem., 2009, 44, 5023-5028.
- P.Y. Marrero, G.J.A. Castillo, F. Torrens, Z.V. Romero, E.A. Castro, Molecules., 2004, 9(12), 1100-1123.
- T. Stams, S. Niranjanakumari, C.A. Fierke, D.W. Christianson, Science., 1998, 280, 752-755.



