Research Article - Der Pharma Chemica ( 2017) Volume 9, Issue 2
Effect of Na, K, Ca, Mg, Fe and Zn on the Structure and Physical Parameters of Protein
Hadeer KE Mohamed1, Sara M Moatmed1, Gehad AR Mabrouk1, Sara M Foly1, Amal S Abd El-Aal1, Amani S Abd el kareem1, Zeinab E-E Abd Elazem1, Fatma I Ali1, Rahma A Abdelhay1, Romissaa M Mohammed1, Alyaa S Garhy1, Nada A Ismail1, Rana A Sayed1, Suzan E Abd elhafiz1 and Medhat Ibrahim22Spectroscopy Department, National Research Center, 12622, Dokki, Giza, Egypt
Abstract
The amino acid alanine is choosen as a model molecule for protein structure. To study the effect of metals upon protein Na, K, Ca, Mg, Fe and Zn were respectively coordinated to alanine through the H-bonding of Carboxyl group. Both Na and K are coordinated with one alanine unit while Ca, Mg, Fe and Zn are coordinated with two alanine units. Calculations are conducted with density functional theory at B3LYP/6-31g(d,p) level of theory. The geometrical parameters are calculated including bond lengths and bond angle of COOH group. The total dipole moment, HOMO/LUMO band gap energy and C=O vibration of COOH group are also calculated. Results indicate that, the bond length LC-O is decreased while LC=O and LO-H are increased, a shift in the characteristic band of carboxyl group (C=O) toward lower wavenumbers is recorded. The total dipole moment and band gap energy are changed. It could be concluded that the studied elements are changing both the structural, Physical and vibrational characteristics of protein.
Keywords
Protein, Alanine, COOH, B3LYP/6-31g(d,p)
Introduction
Although semiempirical methods are fast there are other approximate quantum mechanical methods which were developed to study organic molecules. The parameters of this model are calculated from density functional theory and known as Self-Consistent Charge and Density-Functional Tight-Binding (SCCDFTB) method [1-3]. Amino acids are special class of organic molecules. There are 20 different standard L-α-amino acids used by cells for protein construction. Amino acids, contain both a basic amino group and an acidic COOH group [4]. Alanine is one of the simplest α-amino acids, it is classified as an aliphatic amino acid. It is worth to mention that, the methyl group of alanine never directly interfered in protein function [5-8]. Both experimental and DFT calculations were conducted to study molecular structure and vibrational spectra of nonlinear optical crystal L-alanine cadmium chloride [9].
Carboxyl group are one of the most important functional group which dedicate it for many studies [10,11]. Many structures containing carboxyl group were subjected to both theoretical and experimental study [12].
Molecular modeling was supporting FTIR to study the molecular structure of protein [13]. The effect of heavy metals on protein structure was elucidated with molecular modeling calculations and FTIR spectroscopy [14]. Interaction between metal and amino acids continue to be a topic of research work worldwide according to its wide range of applications [15-17].
In the present work B3LYP/6-31g(d,p) was utilized to optimize alanine then alanine interacted with Na, K, Ca, Mg, Fe and Zn respectively. The total dipole moment, HOMO/LUMO band gap energy and vibrational characteristics were calculated at the same level of theory.
Computational Details
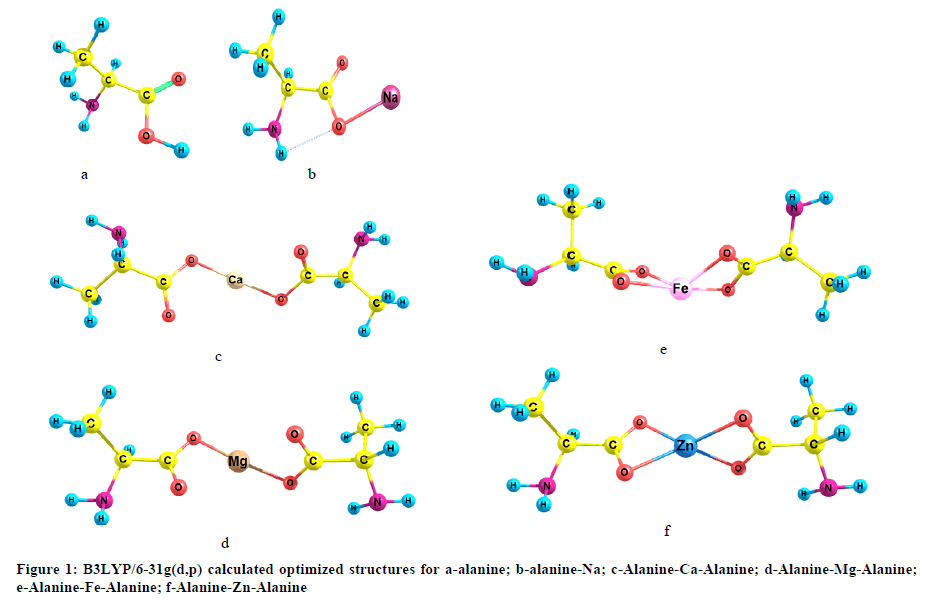
The geometries of alanine; alanine-Na; Alanine-Ca-Alanine; Alanine-Mg-Alanine; Alanine-Fe-Alanine and Alanine-Zn-Alanine were built as indicated in Figure 1. The studied model molecules were optimized with Gaussian 09 soft code at Spectroscopy Department, National Research Centre [18]. The calculations were conducted with density functional theory at B3LYP/6-31g(d,p) quantum mechanical level [19-21]. Vibrational spectra were calculated at the same level of theory.
Conclusion
Monovalent and divalent elements are supposed to interact with alanine through the hydrogen bonding of COOH. Although Ca, Mg, Fe and Zn are divalent but their cooridation with the two alanine units are different. Both Ca and Mg show bidentate coordination while Fe and Zn shoe bidentate cooridation. The geometry of the studied amino acid is changed as one bond length is decreased (LC-O) while other bond lenthgs are increased (LC=O and LO-H) with a change in the bond angle C-O=C. The change in the geometrical parameters are followed with a shift in the characteristic band of carboxyl group which shift the C=O toward lower wavenumbers. Both the total dipole moment and HOMO/LUMO band gap energy are changed as a result of interaction between the studied metals and the studied amino acid.
References
[1] M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai, G. Seifert, Phys. Rev., B, 1998, 58, 7260.
[2] M. Elstner, D. Porezag, G. Jungnickel, T. Frauenheim, S. Suhai, G. Seifert, Mater. Res. Soc., Pittsburgh, 1998, 131.
[3] M. Elstner, T. Frauenheim, E. Kaxiras, G. Seifert, S. Suhai, Phys. Stat. Sol., 2000, 217, 357.
[4] T.G. Cooper, G.M. Day, W. Jones, W.D.S. Motherwell, Acta Cryst., 2006, 62, s229.
[5] N. Grosser, S. Oberle, G. Berndt, K. Erdmann, A. Hemmerle, H. Schröder, Biochem. Biophys. Res. Commun., 2004, 314, 351.
[6] K.S. Kumar, T. Raghavalu, V. Mathivanan, M. Kovendhan, B. Sivakumar, G.R. Kumar, S.G. Raj, R. Mohan, J. Cryst. Growth., 2008, 310, 1182.
[7] G.M. Day, T.G. Cooper, Cryst. Eng. Comm., 2010, 12, 2443.
[8] A.H. Reshak, G. Lakshminarayana, H. Kamarudin, I.V. Kityk, S. Auluck, J. Berdowski, Z. Tylczynski, J. Mater. Sci. Mater. El., 2012, 23, 1922.
[9] I. Cicili Ignatius, S. Dheivamalar, K. Kirubavathi, K. Selvaraju, Int. J. Comp. Mat. Sci. Eng., 2016, 05, 1630001.
[10] M. Ibrahim and E. Koglin, Acta Chim. Slov., 2004, 51(3), 453.
[11] M. Ibrahim J. Comput. Theor. Nanosci., 2009, 6(3), 682.
[12] M. Ibrahim, A. Nada, D-E. Kamal, Indian J. Pure Appl. Phy,, 2005, 43, 911.
[13] Z. Al-Fifi, M. Eid, M. Ibrahim, J. Comput. Theor. Nanosci., 2013, 10, 2381.
[14] H. Elhaes, N.M. Elkashef, F.Kh. Abdel-Gawad, A.M. Shaban, M. Ibrahim, J. Comput. Theor. Nanosci., 2014, 11(4) 1081.
[15] S. Mandal, G. Das, H. Askari, J. Mole. Struct., 2015, 1100, 162.
[16] M. Remelli, V.M. Nurchi, J.I. Lachowicz, S. Medici, M.A. Zoroddu, M. Peana, Coord, Chem. Rev., 2016, 55, 327-328.
[17] X.Y. Xu, B. Yan, Sensor Actuat. B. Chem., 2016, 230, 463.
[18] M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, Inc., Wallingford, CT, 2010.
[19] A.D. Becke, Chem. Phys., 1993, 98, 5648.
[20] C. Lee, W. Yang, R.G. Parr, Phys. Rev., 1998, 37(2) 785.
[21] B. Miehlich, A. Savin, H. Stoll, H. Preuss, Chem. Phys. Lett., 1989, 157(3), 200.
[22] M. Ibrahim, H. El-Haes, Int. J. Environ. Pollut., 2005, 23(4), 417.
[23] M. Ibrahim, A.A. Mahmoud, J. Comput. Theor. Nanosci., 2009, 6, 1523.



