Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 3
Electrochemical Surface Oxidation of Graphite Electrode and Its Superior Sensitive Platform for Endocrine Disrupting Compounds
Ravindra B Kohakade1, Senthil Kumar1, Raghu S Bose2 and Raman A Kalaivani1*
1Department of Chemistry, Vels University, Pallavaram, Chennai-600117, Tamilnadu, India
2Department of Vels Advanced Energy Research Centre, Vels University, Pallavaram, Chennai-600117, Tamilnadu, India
- *Corresponding Author:
- Raman A Kalaivani
Department of Chemistry
Vels University
Pallavaram, Chennai-600117, Tamilnadu, India
Abstract
Here we first time report, the investigation of stable surface (2 mm) oxidized graphite electrode prepared via the anodic oxidation of graphite in different electrolyte medium such as KOH, H2SO4 and KH2PO4. In the electrochemical surface oxidation route, high quality graphene oxide was produced on surface of the graphite rod. The formation of oxidized graphite (Graphene oxide) was proved by X-ray Diffraction (XRD) and Raman spectroscopy analysis. The synthesis product was also characterized by Scanning Electron Microscopy (SEM), etc. The surface oxidized graphite electrode was used for the simultaneous determination of 692 mV catechol and 1.31 V methylparaben. Unbuffered 0.1 M Na2SO4 was used as Supporting Electrolyte (SE), as hydro-alcoholic solution (in 1/4 ethanol/water volume ratio, i.e., 20% volume of ethanol in the final working solution from the cell). The fast and sensitive electrochemical responses for the detection of catechol and methylparaben at a potential between 0 to 1.5 V were obtained. The cyclic voltammetric measurements yielded calibration plots with very superior linearity and high sensitivity Results demonstrated that the electrochemically surface modified graphite electrode displayed a good performance along with high sensitivity and long-term stability.
Keywords
Catechol, Methylparaben, Electrochemically surface oxidized graphite electrode
Introduction
In beauty products processing, drugs and new food technology, the chemical preservation has become an important practice, with addition of commonly used synthetic preservatives developed from benzoic acid and hydroxybenzoic acid as alkyl para (4)-hydroxybenzoates, so-called parabens or alkyl parabens (methyl, ethyl, etc.) and electron-donating alkyl gallates, derivatives of gallic acid, as antioxidants [1-10]. Among many additives, synthetic and naturally occurring compounds are used, especially as antioxidants. Electrochemistry of various natural antioxidants is the subject of an active research as is the electrochemical study of the phenolic compounds or their derivatives [11-20]. However, there are relatively few recent papers focused on the electrochemical study of the used preservatives from the parabens series, employed in food, pharmaceutical or cosmetic products, alone or in combination with some associated antioxidant additives from the alkylgallates series.
It also has negative biological effect and possible side effects on humans and other mammals, such as estrogenic effects which may involve breast cancer risk [21-25]. Study of such effects requires correlation with their water solubility, or lipophilicity and toxicity characteristics. In this overall context, the relationships between structure and activity, the composition Sensors and stability of the compounds, and their dosage in target applications and frequency of their use, are especially important. In this work, the electrochemical behaviour of some preservatives-antioxidants methyl parabens, catechol was investigated in hydro-alcoholic and aqueous solutions, at a electrochemically oxidized surface of graphite rod. Especially in the anodic potential range. To the best of our knowledge, no anodic sensing, characterization and determination of these commonly used preservatives-antioxidant food additives at a electrochemically oxidized surface of graphite rod was made.
The principal aim of this work, which has an introductory character, was to test and collect new data for the simultaneous electrochemical determination of catechol and methylparaben using electrochemically oxidized surface of graphite rod.
Catechol (CC) can induce DNA damage and cause human cancer [26]. Both of these two compounds have been included in the lists of priority pollutant by international bodies, such as the US Environmental Protection Agency (EPA) and the European Union (EU) [27,28]. Therefore, it is highly urgent and necessary to develop a facile, robust, inexpensive, and effective analytical method for the determination of CC and methylparaben with high sensitivity and selectivity. Many methods have been used for catechol and determination, such as high performance liquid chromatography, fluorescence, gas chromatography/mass spectrometry and capillary electrochromatography [29-34]. Among those methods, electrochemical assays have attracted increasing attention due to their great promise for portability, low cost and high sensitivity [35,36]. Various electrochemical sensors have been developed for the phenol compounds sensitive detection [37-41]. Nanomaterials, such as graphene, carbon nanotubes and metal-oxidation nanoparticles [42-44], have been modified on electrode surface to enhance the sensitivity of the sensors. Besides, different polymers have been employed to prepare electrochemical sensors for CC and determination because of their high electrocatalytic activity. At the same time, selectivity is also an important property of these sensors due to the high over potentials of phenol compounds without a separation between their oxidation peaks and the coexistence of a variety of organic compounds [45,46].
Formulae of the investigated compounds are
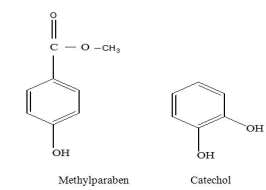
Experimental Section
Chemicals and reagents
Methylparaben, catechol, sodium sulphate, ethanol, disodium hydrogen phosphate and sodium hydrogen phosphate were used directly. These reagents were purchased from Sigma Aldrich. Graphite rod of 2 mm surface was purchased from Merck. Doubled distilled water was used to prepare 0.1 M buffer solution. All electrochemical experiments were carried out at room temperature 25 ± 0.1°C.
Instrument
The electrochemical measurements were carried out in an undivided Metrohm three electrode cell equipped with an electrochemically oxidized graphite rod, a (2 mm) diameter as working electrode, a platinum foil counter electrode and a Saturated Calomel Electrode (SCE) as reference. The graphite rod was parched from Merck. The working electrode, used in our previous papers was performed and stabilized by subjecting it to several hundred cycles between large potential limits in neutral media. The electrochemical data resulted as Cyclic Voltammograms (CVs) and Chronoamperograms (CAs), Linear Swip Voltammetry (LSV), Dispersion Plus Voltammetry (DPV) were obtained by using an CH I760D electrochemical workstation (Chenhua Instrument, Shanghai, China) with a conventional three-electrode system, controlled by a PC.
Here we first time report, the synthesis of stable surface oxidized graphite electrode in phosphate electrolyte medium and its superior catalytic behavior of CC and methylparaben (Figure 1 and Scheme 1). We fabricated electrochemically surface oxidized graphite electrode via electrochemical oxidation of graphite and self-assembly method. The as-prepared electrochemically surface oxidized graphite electrode was characterized with unique structural features and electrochemical properties, which provided it excellent performance as a promising electrode material. Compared with the bare, modified electrode exhibited much higher electrocatalytic activities toward the oxidation of CC and methylparaben. Hence, the modified electrode based electrochemical sensor was used for the simultaneous determination of CC and methylparaben with good selectivity and high sensitivity.
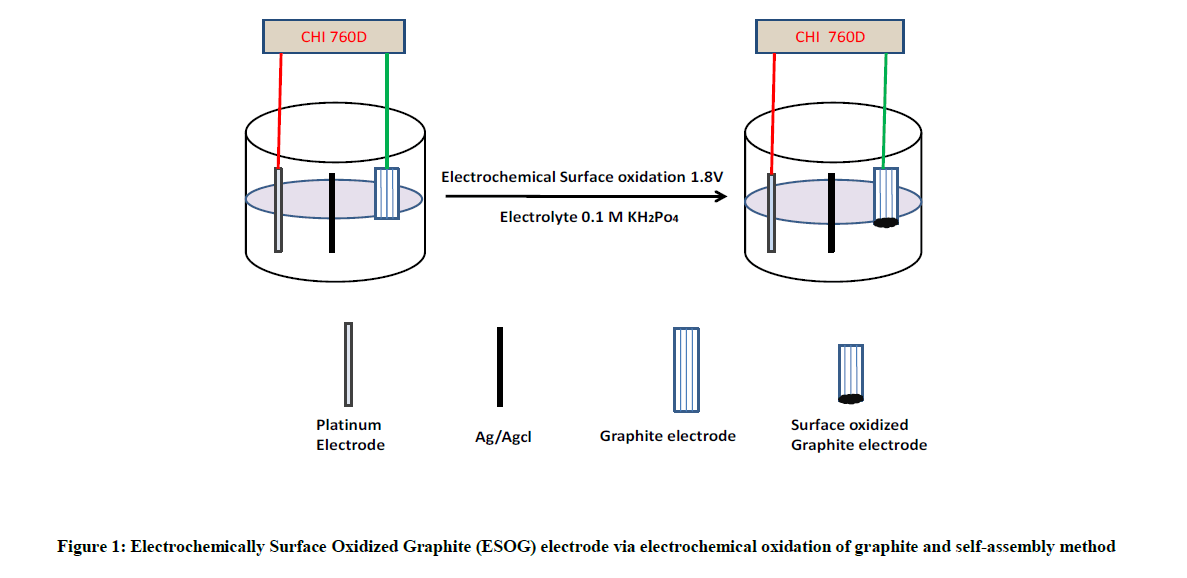
Figure 1: Electrochemically Surface Oxidized Graphite (ESOG) electrode via electrochemical oxidation of graphite and self-assembly method
From LSV scanning from 0 to 1.8 V of three different electrolytes, i.e. KOH, H2SO4, KH2PO4, we studied that potassium dihydrogen phosphate is mild oxidizing agent and does not exfoliate, only surface modification takes place, so we have taken KH2PO4 as electrolyte. The proposed electrochemical oxidation mechanism showed that PO4- and H2O can be oxidized to form surface oxidized graphite rod, surface oxidized graphite rod was obtained by the formation of gaseous H2 and O2 within graphite rod. The graphite rod (Anode) and platinum (Cathode) were placed vertically at bottom and top of the electrochemical cell, with KH2PO4 solution as electrolyte.
Characterization
Bruker AXS D8 Advance X-ray Diffractometer (XRD) was used for X-ray Powder Diffraction (XRPD) analysis of the synthesized samples. D8 most Advance system was prepared with a 2.2 kW Cu anode X-ray tube and a Lynx Eye position sensitive detector. All powder patterns were obtained by Cu Kα radiation (λ=1.5406 A). Scanning Electron Microscope (Hitachi S-3400 N) were, used to observe morphology and elemental composition of the synthesized graphene. Perkin Elmer BX FT-IR spectrometer in the range of 4000-400 cm-1 was used to record Fourier Transform Infrared (FTIR) spectra of samples.
Fabrication of modified electrode
We fabricated modified electrode via electrochemical oxidation of graphite and self-assembly method.
Results and Discussion
Possible mechanism of electrochemically surface oxidized graphite electrode
From linear sweep voltammetry scanning from 0 to 1.8 V of three different electrolyte, i.e., KOH, H2SO4, KH2PO4, were studied that Potassium dihydrogen phosphate is mild oxidizing agent and does not exfoliate, only surface modification takes place, so we have taken KH2PO4 as electrolyte (Figure 2).
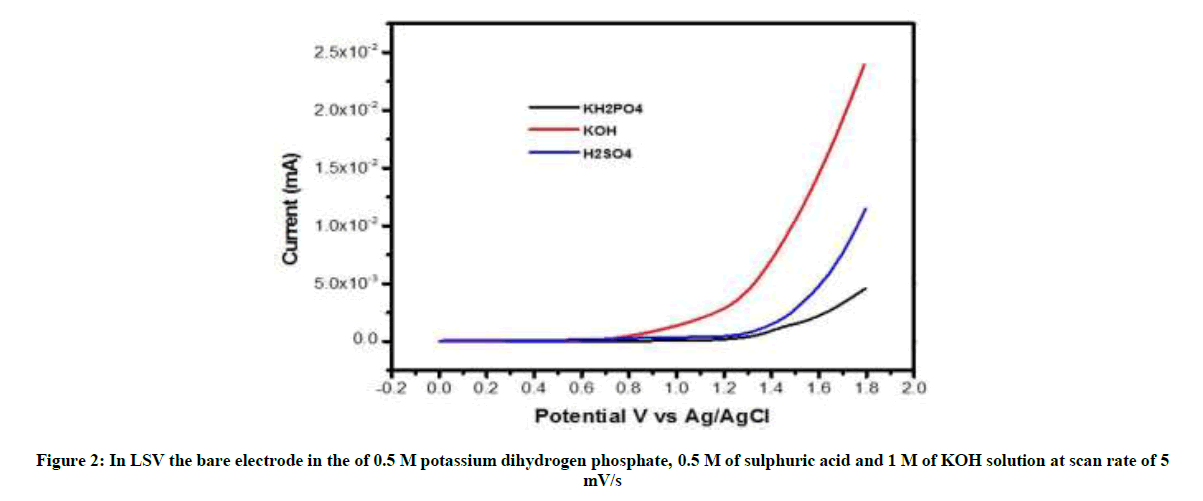
Figure 2: In LSV the bare electrode in the of 0.5 M potassium dihydrogen phosphate, 0.5 M of sulphuric acid and 1 M of KOH solution at scan rate of 5 mV/s
Synthesis of stable surface (2 mm) oxidized graphite electrode via the anodic oxidation of graphite inKH2PO4 solution, short time at 1 h. The proposed electrochemical oxidation mechanism showed that PO4- and H2O can be oxidized to form surface oxidized graphite rod, surface oxidized graphite rod was obtained by the formation of gaseous H2 and O2 within graphite rod.
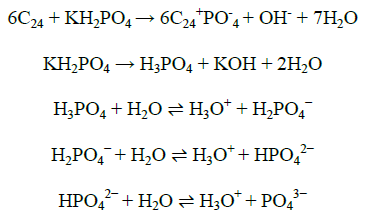
Possible mechanism of electrochemical oxidation: (i) Oxidation at the edging sites as well as grain borders which helps to depolarization and extension of the graphite layers, thus facilitates the oxidation of phosphate (PO42-) anions inside the graphitic layers. At some time, co-intercalation of H2O molecule with PO42− anions. (ii) Self-oxidation of water and Phosphate (PO42-) anions create gaseous species such as P2O5, O2 and etc., as proof for release of vital gas during electrochemical process.
Chemical structure of electrochemically surface oxidized graphite rod was confirmed by FTIR spectral analysis in attendance of different oxygen functionalities, (Figure 3a) which have been proved, the band at 1220 cm−1 corresponds with the C-O in the epoxide group, the bands at 1720 and 1618 cm-1, correspond to the C=O carbonyl/carboxyl and C=C aromatic groups respectively and the band at 3420 cm-1, corresponds to the O-H group. For the ESOG, Figure 3b shows the O-H band at 3430 cm−1 was oxidized in intensity due to the oxygenated functionalities. The XRD patterns of electrochemically surface oxidized of graphite were characteristic (Figure 3b) Confirmation of the electrochemically surface exfoliation of graphite and formation of graphite oxide is proved with peak at 2θ=25.54°, with a d-spacing of 0.34 nm.
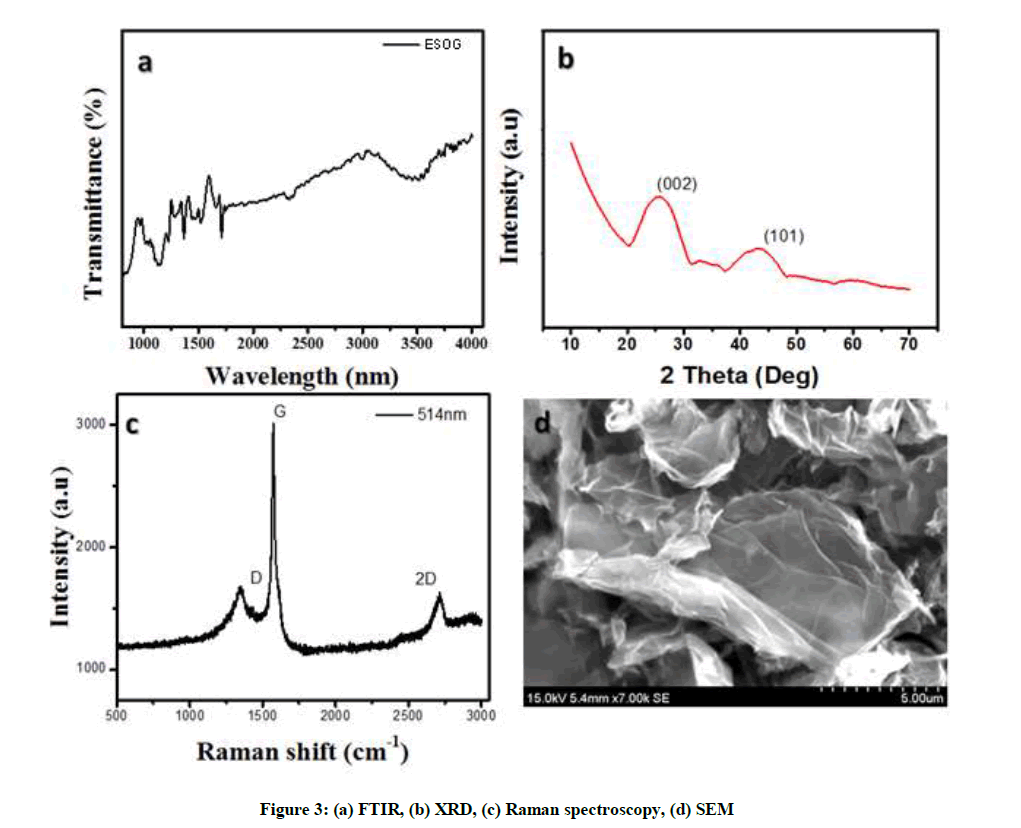
Figure 3: (a) FTIR, (b) XRD, (c) Raman spectroscopy, (d) SEM
The Raman spectrum of electrochemically surface oxidized of graphite (Figure 3c) The most important Raman features of the modified electrode are, G band (1547 cm-1), D band (1351 cm-1). The small concentration D peak in the spectrum near 1346 cm-1 shows the soaring superiority and little defects. The appearance of the D peak could be accredited to the disorder in the carbon lattice such as the edging of domain and domain borders. The widening of Bragg’s peak indicates the formation of crystalline graphite oxidized whose size is calculated using the Scherer formula, Equation 1:
D = Kλ/βs cosθ ------- (1)
Where, D is the average size of the crystal, K is the shape dependent Scherer’s constant, wavelength of radiation is λ, full peak width at half maximum is βs and θ is the diffraction angle. Using the full Width Half Maxima (FWHM) of (111) peak, crystal sizes of the ESOG calculated are 2.01 nm. The crystal size calculated from ID/IG ratio using the formula, Equation 2:
La = 2.4 × 10-10λ4 [ID/IG]-1 -------- (2)
Where, La is 3.8 nm respectively. Scanning Electron Microscope (SEM) was used for studying surface morphology of the prepared modified electrode, Flaky texture of modified electrode was obtained by incomplete exfoliation. This suggests that incomplete exfoliated structure and reflects its coated microstructure huge inter-layer spacing and bulky multi-layer stacks, as noted in literature. Figure 3d shows the Scanning electron microscope images of electrochemically surface oxidized graphite oxide respectively. From figures it shows that, the surface oxide graphite oxidizes sheets were translucent as well as wrinkly in environment. This wrinkled graphite oxide is very advantageous to maintain a elevated surface area of the electrode since the sheets cannot readily collapse back to a graphitic structure consisted of randomly aggregated thin crumpled sheets that are strongly associated within them self, for formation of disordered solid.
Ethanol and water were used as solvents for standard alcoholic or saturated aqueous stock solutions, and unbuffered 0.1 M Na2SO4 as Supporting Electrolyte (SE), as hydro-alcoholic solution (in 1/4 ethanol/water volume ratio, i.e., 20% volume of ethanol in the final working solution from the cell). The use of ethanol was selected for compatibility and to prevent a separation from the solution of the target analyte and an advanced fouling of the electrode through reactants or reaction products. The working solution in the cell (50 ml) has been prepared freshly from a more concentrated Na2SO4 aqueous solution diluted adequately with water and ethanol in the above mentioned volume ratio. By adding a very small volume of the relatively concentrated samples in the large volume of the working solution from the cell, the modification in the overall ethanol content was insignificant.
The CV, LSV, DPV measurements were accomplished using 50 ml volumes of solutions in the working cell and predominantly an unbuffered hydroalcoholic neutral media described above. A 0.05 Vs-1 scan rate was used predominantly. Working temperature was 23 ± 100°C. The CV, LSV, DPV investigations occurred predominantly at neutral pH.
Cyclic voltammetry data in hydro-alcoholic solutions
Figure 1 depicts as examples, typical CVs involving successive first and second scan-curves 1 and 2, with reference to the supporting electrolyte with the accessible potential window, and the presence of CC and methylparaben at a relative high scan rate.
Figure 4 shows cyclic voltammogram recorded in the individual solutions. The electrochemical behavior of surface oxidized graphite electrode was first tested with cyclic voltammetry for its response to catechol and methylparaben figure shows cyclic voltammograms at a different scan rate. The oxidation peak current of catechol and methylparaben were recorded.
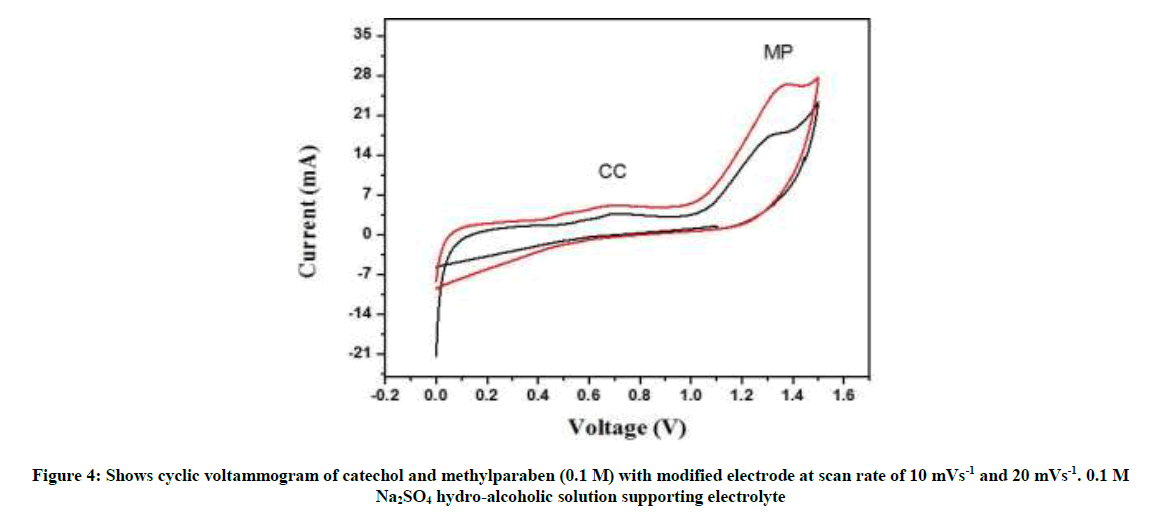
Figure 4: Shows cyclic voltammogram of catechol and methylparaben (0.1 M) with modified electrode at scan rate of 10 mVs-1 and 20 mVs-1. 0.1 M Na2SO4 hydro-alcoholic solution supporting electrolyte
Redox reaction of CC at electrochemically oxidized graphite rod should be a two-electron and two-proton process. And the probable oxidation reactions of CC on electrochemically oxidized graphite rod are described as follows.
Effect of scan rate
The effect of scan rate on the electrochemical behavior of catechol and methylparaben was studied on the electrochemically oxidized graphite rod (surface) by CV method. As shown in Figure both the redox peak currents of catechol and methylparaben increased with increasing scan rate from 10 to 20 mV s-1 (Figures 5 and 6).
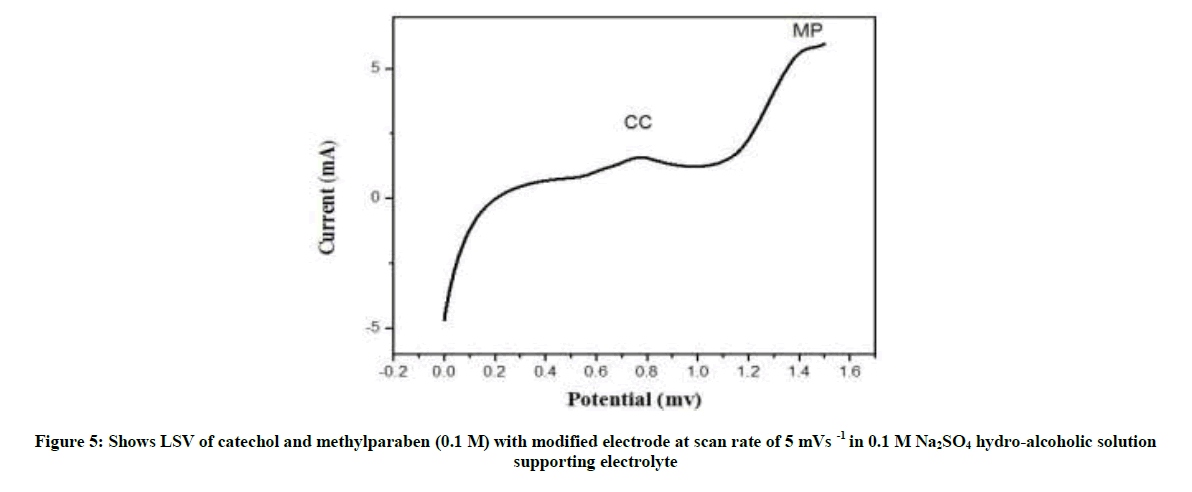
Figure 5: Shows LSV of catechol and methylparaben (0.1 M) with modified electrode at scan rate of 5 mVs-1 in 0.1 M Na2SO4 hydro-alcoholic solution supporting electrolyte
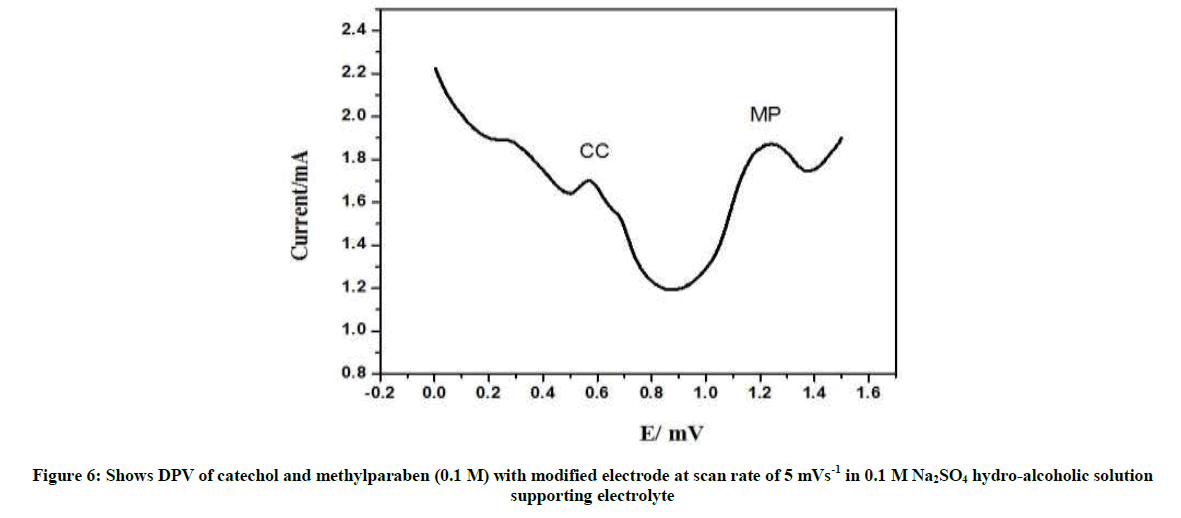
Figure 6: Shows DPV of catechol and methylparaben (0.1 M) with modified electrode at scan rate of 5 mVs-1 in 0.1 M Na2SO4 hydro-alcoholic solution supporting electrolyte
Conclusions
Electrochemical study of and CC and methylparaben (Endocrine disrupting compounds), the adequate selection of the supporting electrolyte or the aspects regarding the presence and concentration of the investigated compounds in the aqueous system were advantageously accomplished by the use of electrochemically modified surface of graphite rod. The electrochemically modified surface of graphite electrode is very useful for the study of the electrochemistry of CC and methylparaben; the initial steps in the development of the several electroanalytical alternatives were tested. The CV, LSV and DPV measurements yielded calibration plots with very good linearity and sensitivity, useful for detection and analytical applications. The electrochemically modified surface of graphite rod offered a sustainable basis for the present and further electroanalytical development and application.
References
- E.I. Korotkova, O.A. Avramchik, T.M. Angelov, Y. Karbainov, Electrochimica Acta., 2005, 51, 324.
- S.H. Kang, H. Kim, J. Pharm. Biomed. Anal., 1997, 15, 1359.
- S. Gunckel, P. Santander, G Cordano, J. Ferreira, S. Munoz, L.J. Nunez-Vergara, J.A Squella, Chem. Bio. Interact., 1998, 114, 45.
- B. Saad, Md. F. Bari, M.I. Saleh, K. Ahmad, M.K. Tolib, J. Chromaogr., 2005, 1073, 393.
- Q. Zhang, M. Lian, L. Liu, H. Cui, Anal. Chim. Acta., 2005, 537, 31.
- F. Croo, J. Schutter, W. Bossche, P. Moerloose, Chromatographia., 1984, 18, 260.
- T.G. Diaz, A.G. Cabanillas, M.F.A. Franco, F. Salinas, J.C. Vire, Electroanalysis., 1998, 10, 497.
- M. Szymula, M.J. Narkeviccz, J. Appl. Electrochem., 2006, 36, 455.
- E. Diego, L. Agui, A. Gonzales, P. Yanez-Sedeno, J.M. Pingarron, J.M. Kauffmann, Electroanalysis., 1998, 10, 33.
- M.D. Morales, M.C. Gonzales, A.J. Revejo, J.M. Pingarron, Microchem. J., 2005, 80, 71.
- C. Ceballos, H. Fernandez, J. Braz. Chem. Soc., 1995, 6, 1.
- S.C. Litescu, G.L. Radu, Tecnol., 2000, 211, 218.
- S.C. Litescu, N. Cioffi, L. Sabbani, G.L. Radu, Electroanalysis., 2002, 14, 858.
- I. Castro-Gamboa, C.L. Cardoso, D.H.S. Silva, A.J. Cabalheiro, M. Furlan, V. Silva Bolzani, J. Braz. Chem. Soc., 2003, 14, 771.
- C. Giacomelli, F.C. Giacomelli, L.O. Alves, A.K. Timbola, A. Spinelli, J. Braz. Chem. Soc., 2004, 15, 748.
- A.K. Timbola, C.D. Souza, C. Giacomelli, A. Spinelli, J. Braz. Chem. Soc., 2006, 17, 139.
- C. Radovan, D. Cinghită, F. Manea,. M. Mincea, C. Cofan, V. Ostafe, Electrode Sensors., 2008, 8, 4330.
- D. Sopchak, B. Miller, Y. Avygal, R. Kalish, J. Electroanal. Chem., 2002, 538, 39.
- R. Ramesham, M.F. Rose, J. Mat. Sci. Lett., 1997, 16, 799.
- J. Wang, R.P. Deo, M. Musameh, Electroanalysis., 2003, 15, 1830.
- C. Lemini, R. Jaimez, M.E. Avila, Y. Franco, F. Larrea, A.E. Lemus, Toxicol. Ind. Health., 2003, 19, 69.
- C. Lemini, A. Hernandez, R. Jaimez, Y. Franco, M.E. Avila, A. Castell, Toxicol. Ind. Health., 2004, 20, 123.
- S. Oishi, Food Chem. Toxicol., 2002, 40, 1807.
- E.J. Routledge, J. Parker, J. Odum, J. Ashby, J.P. Sumper, Toxicol. Appl. Pharm., 1998, 153, 12.
- P.W. Harvey, J. Appl. Toxicol., 2003, 23, 285.
- S. Kawanishi, Y. Hiraku, M. Murata, S. Oikawa, Free Radic. Biol. Med., 2002, 32, 822.
- A.J. Ahammad, S. Sarker, M.A. Rahman, J.J. Lee, Electroanalysis., 2010, 22, 694.
- A.J. Ahammad, M.M. Rahman, G.R. Xu, S. Kim, J.J. Lee, Electrochim. Acta., 2011, 56, 5266.
- C. Terashima, T.N. Rao, B.V. Sarada, D.A. Tryk, A. Fujishima, Anal. Chem., 2002, 74, 895.
- D.S. Bhatkhande, V.G. Pangarkar, A.A. Beenacker, J. Chem. Technol. Biot., 2002, 77, 102.
- M.V. Bosco, M. Garrido, M.S. Larrechi, Anal. Chim. Acta., 2006, 559, 240.
- A. Afkhami, H.A. Khatami, J. Anal. Chem., 2001, 56, 429.
- S.C. Moldoveanu, M. Kiser, J. Chromatogr. A., 2007, 1141, 90.
- C. Nilsson, S. Nilsson, Electrophoresis, 2006, 27, 76.
- F. Li, X. Han, S. Liu, Biosens. Bioelectron., 2011, 26, 2619.
- S. Liu, Y. Wang, C. Zhang, Y. Lin, F. Li, Chem. Commun., 2013, 49, 2335.
- H. Yin, Q. Zhang, Y. Zhou, Q. Ma, L Zhu, S. Ai, Electrochim. Acta., 2011, 56, 2748.
- L. Chen, Y. Tang, K. Wang, C. Liu, S. Luo, Electrochem. Commun., 2011, 13, 133.
- D.M. Zhao, X.H. Zhang, L.J. Feng, L. Jia, S. Wang, Colloids Surf. B., 2009, 74, 317.
- J. Yu, W. Du, F. Zhao, B. Zeng, Electrochim. Acta., 2009, 54, 984.
- Y.J. Yang, L. Weikun, Fullerenes, Nanotubes, Carbon Nanostruct., 2015, 23, 410.
- Q. Guo, J. Huang, P. Chen, Y. Liu, H. Hou, T. You, Sens. Actuators B., 2012, 163, 179.
- S. Chandra, H. Lang, D. Bahadur, Anal. Chim. Acta., 2013, 795, 8.
- K.J. Huang, L. Wang, J. Li, M. Yu, Y.M. Liu, Microchim. Acta., 2013, 180, 751.
- M.U.A. Prathap, B. Satpati, R. Srivastava, Sens. Actuators. B., 2013, 186, 67.
- D. Lakshmi, A. Bossi, M.J. Whitcombe, I. Chianella, S.A. Fowler, S. Subrahmanyam, E.V. Piletska, S.A. Piletsky, Anal. Chem., 2009, 81, 3576.



