Research Article - Der Pharma Chemica ( 2017) Volume 9, Issue 1
Facile Synthesis of m-Terphenyls by Ring Transformation of 2H-Pyran-2-ones
Priyanka Kole, Samata Shetgaonkar and Fateh V Singh*Fateh V Singh, Chemistry Division, School of Advanced Science, VIT University, Chennai-600127, Tamilnadu, India,
Abstract
An alternative approach for the synthesis of meta-terphenyls 3a-g functionalized with electron-withdrawing or -donating groups is described and illustrated by carbanion-induced ring transformation of 6-aryl-2H-pyran-2-ones 1a-g with propiophenone 2 in excellent yields.
Keywords
2H-Pyran-2one, Ring transformation reaction, Nucleophile, Terphenyls
Introduction
The chemistry of terphenyls has been developed as an important research area in organic chemistry. Terphenyls bearing electron donating and accepting functionalities are the key components several mushrooms belonging to the Thelephoraceae family [1-4]. Theleforin A 1 was isolated from fruiting bodies of the mushroom Thelephora vialis and exhibits antioxidant activity [5]. Furthermore, atromentin 2 was isolated from methanolic extract of fruiting bodies of the mushroom Thelephora aurantiotincta [6]. Terprenin 3 was obtained from the fermentation broth of Aspergillus candidus RF-5672 [7-9] and identified as potent immunoglobulin E antibody suppressant [10,11]. There are several other terphenyl-cored naturally occurring compounds reported in past decades and known for diverse biological properties (Figure 1) [6-13].
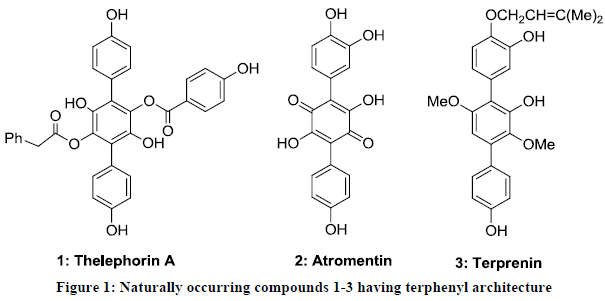
Various synthetic terphenyls have been proved their importance in medicinal and pharmaceutical chemistry. Few terphenyl derivatives have been designed as selective inhibitors for dihydroortate dehydrogenase [14] and cyclooxygenase enzymes [15]. In addition, few synthetic terphenyls have been developed as potent antidiabetic agents [16-18].
Materials and Methods
Melting points were obtained in open capillary tubes. 1H NMR and 13C NMR spectra were recorded on a AV-400 Bruker using the solvents indicated with 400 and 100 MHz, respectively, also a DRX-500 Bruker was used in the some cases for 1H (500 MHz) and 13C (125 MHz). Mass spectra (m/z) were recorded under the conditions of electron impact (EI) and electrospray (ES) and chemical
ionization (CI). All reactions were monitored by thin-layer chromatography that was performed on pre-coated sheets of silica gel 60, and column chromatography was performed with silica gel 60 (Avra synthesis, 100–200 mesh). Eluting solvents are indicated in the text. Dry THF was used from a solvent purification system while acetonitrile of HPLC grade was used and dried with molecular sieves (4 Å). All other purchased chemicals were used without further purification.
Experimental Section
General procedure for the synthesis of compounds 3a-g
A mixture of 6-aryl-2H-pyran-2-one (1) (1.0 mmol), propiophenone (2) (1.2 mmol) and powdered KOH (1.2 mmol) in dry DMF (5 mL) was stirred at room temperature for 10-15 h. The course of reaction was monitored with TLC. After the completion of reaction, the reaction mixture was poured into ice water with vigorous stirring and finally neutralized with dilute HCl. The solid thus obtained was filtered and purified on a neutral alumina column using EtOAc-hexane (1:9) as eluent. The isolated compounds were characterized as m-terphenyls 3 by their spectroscopic analysis.
2'-Methyl-5'-(piperidin-1-yl)-[1,1':3',1''-terphenyl]-4'-carbonitrile 3a
White solid; mp 112-114ºC; 1H NMR (400 MHz, CDCl3) δ 2.38 (s, 3H, Me), 1.58-1.68 (m, 2H, CH2), 1.75-1.90 (m, 4H, 2CH2), 3.00-3.20 (m, 4H, 2CH2), 6.88 (s, 1H, ArH), 6.91-7.02 (m, 4H, ArH), 7.06-7.23 (m, 6H, ArH); IR (KBr) 2210 cm-1 (CN); MS (ESI) 353 (M++1).
4-Fluoro-2'-methyl-5'-(piperidin-1-yl)-[1,1':3',1''-terphenyl]-4'-carbonitrile 3b
White solid; mp 132-134ºC; 1H NMR (400 MHz, CDCl3) δ 2.37 (s, 3H, Me), 1.60-1.69 (m, 2H, CH2), 1.76-1.90 (m, 4H, 2CH2), 3.03- 3.22 (m, 4H, 2CH2), 6.91 (s, 1H, ArH), 6.88-7.03 (m, 4H, ArH), 7.12 (d, J = 8.4 Hz, 2H, ArH), 7.17-7.24 (m, 3H, ArH); 13CNMR (100 MHz, CDCl3) 25.3, 26.8, 31.9, 105.4, 117.8, 118.9, 127.6, 128.4, 128.6, 130.9, 132.5, 133.7, 134.9, 139.5, 140.3, 145.6, 152.6, 158.5; IR (KBr) 2213 cm-1 (CN); MS (ESI) 471 (M++1).
4-Chloro-2'-methyl-5'-(piperidin-1-yl)-[1,1':3',1''-terphenyl]-4'-carbonitrile 3c
White solid; mp 121-123ºC; 1H NMR (400 MHz, CDCl3) δ 2.38 (s, 3H, Me), 1.56-1.69 (m, 2H, CH2), 1.73-1.91 (m, 4H, 2CH2), 3.01-3.22 (m, 4H, 2CH2), 6.85 (s, 1H, ArH), 6.83-7.02 (m, 4H, ArH), 7.09 (d, J = 8.4 Hz, 2H, ArH), 7.13-7.22 (m, 3H, ArH); 13CNMR (100 MHz, CDCl3) 25.5, 26.7, 31.3, 105.7, 117.8, 118.8, 127.5, 128.3, 128.4, 130.6, 132.4, 133.6, 134.8, 139.5, 140.3, 145.6, 152.3, 158.3; IR (KBr) 2215 cm-1 (CN); MS (ESI) 387 (M++1), 387 (M++3).
4-Bromo-2'-methyl-5'-(piperidin-1-yl)-[1,1':3',1''-terphenyl]-4'-carbonitrile 3d
White solid; mp 140-142ºC; 1H NMR (400 MHz, CDCl3) δ 2.38 (s, 3H, Me), 1.58-1.70 (m, 2H, CH2), 1.70-1.92 (m, 4H, 2CH2), 2.99-3.1 (m, 4H, 2CH2), 6.84 (s, 1H, ArH), 6.88-7.03 (m, 4H, ArH), 7.08 (d, J = 8.4 Hz, 2H, ArH), 7.12-7.24 (m, 3H, ArH); 13CNMR (100 MHz, CDCl3) 25.8, 26.3, 31.7, 106.1, 117.6, 118.3, 127.4, 128.6, 128.7, 130.9, 132.8, 133.3, 134.4, 139.3, 140.6, 145.2, 152.6, 158.8; IR (KBr) 2215 cm-1 (CN); MS (ESI) 431 (M++1), 387 (M++3).
2',4-Dimethyl-5'-(piperidin-1-yl)-[1,1':3',1''-terphenyl]-4'-carbonitrile 3e
White solid; mp 146-148ºC; 1H NMR (400 MHz, CDCl3) δ 2.32 (s, 3H, Me), 2.36 (s, 3H, Me), 1.59-1.66 (m, 2H, CH2), 1.72-1.87 (m, 4H, 2CH2), 3.01-3.20 (m, 4H, 2CH2), 6.72 (d, J = 8.6 Hz, 2H, ArH), 6.82-7.03 (m, 5H, ArH), 7.14-7.24 (m, 3H, ArH); IR (KBr) 2217 cm-1 (CN); MS (ESI) 367 (M++1).
4-Methoxy-2'-methyl-5'-(piperidin-1-yl)-[1,1':3',1''-terphenyl]-4'-carbonitrile 3f
White solid; mp 146-148ºC; 1H NMR (400 MHz, CDCl3) δ 2.38 (s, 3H, Me), 1.58-1.67 (m, 2H, CH2), 1.75-1.89 (m, 4H, 2CH2), 3.00-3.20 (m, 4H, 2CH2), 3.73 (s, 3H, OMe), 6.67 (d, J = 8.6 Hz, 2H, ArH), 6.84-7.01 (m, 5H, ArH), 7.16-7.25 (m, 3H, ArH); 13C NMR (100 MHz, CDCl3) 24.9, 26.7, 32.2, 55.4, 104.7, 116.9, 117.8, 127.2, 128.5, 129.2, 131.5, 133.5, 133.9, 134.7, 138.7, 140.1, 144.8, 152.8, 157.6; IR (KBr) 2214 cm-1 (CN); MS (ESI) 383 (M++1).
3-Methoxy-2'-methyl-5'-(piperidin-1-yl)-[1,1':3',1''-terphenyl]-4'-carbonitrile 3g
White solid; mp 158-160ºC; 1H NMR (400 MHz, CDCl3) δ 2.38 (s, 3H, Me), 1.57-1.67 (m, 2H, CH2), 1.73-1.88 (m, 4H, 2CH2), 2.99-3.18 (m, 4H, 2CH2), 3.76 (s, 3H, OMe), 6.66-6.78 (m, 1H, ArH), 6.83-7.03 (m, 4H, ArH), 7.16-7.25 (m, 5H, ArH); 13C NMR (100 MHz, CDCl3) 24.6, 26.5, 32.4, 55.6, 104.5, 116.7, 117.6, 127.2, 128.4, 128.9, 131.6, 133.4, 133.6, 134.5, 138.6, 140.3, 144.6, 152.5, 157.4; IR (KBr) 2214 cm-1 (CN); MS (ESI) 383 (M++1).
Result and Discussion
Synthesis of m-terphenyl has been achieved by using both classical and non-classical approaches. In classical approaches, m-terpheyls have been synthesized by transition metal-catalyzed cross coupling reactions of electrophilic and nucleophilic reaction partners. In 2015, Larrosa et al. developed Pd-catalyzed synthesis of m-terphenyl by cross-coupling reaction of 1,3-dichloroarene as electrophile and aryl magnesium bromide as nucleophile [19]. Furthermore, Wang et al. achieved the synthesis of m-terphenyls by Pd-catalyzed denitrogenative Hiyama coupling of arylhydrazine with triethoxy(phenyl)silane [20]. Recently, Sandford and et al. developed a direct approach for the synthesis of polyfluorinated m-terphenyls by Pd-catalyzed Suzuki-Miyaura coupling of phenyl
boronic acids with polyfluorinated nitro benzene [21]. There was a solitary approach in which nickel catalyst was used to achieve the synthesis of m-terphenyls [22]. In addition, there are some non-classical approaches used for the synthesis of m-terphenyls including triphyrin-catalyzed reaction of aryliodine with benzene [23], organocatalytic reaction of iodoarene with benzene [24], base-promoted homolytic aromatic substitution 3-chloroiodobenzene with benzene [25] and base-promoted benzannulation reaction 3-formylchromones with 1,3-diphenyl-2-propanone [26]. In addition, ring transformation of 2H-pyran-2one is another non-classical approach that is successfully used for thesis of o-, m- and p-terphenyls [27,28].
The wide-ranging applications and high demand of terphenyls and paucity of non-metal catalyzed synthetic methodologies prompted us to develop a simple, general and efficient route for the synthesis of m-terphenyls. Herein, we report synthesis of functionalized meta-terphenyls through reaction of 2H-pyran-2-ones 1 with propiophenone 2 in high yields at room temperature without using an organometallic reagent or a catalyst. The advantage of the procedure lies in the creation of a central benzene ring under mild reaction conditions from 2H-pyran-2-one at room temperature [29-35].
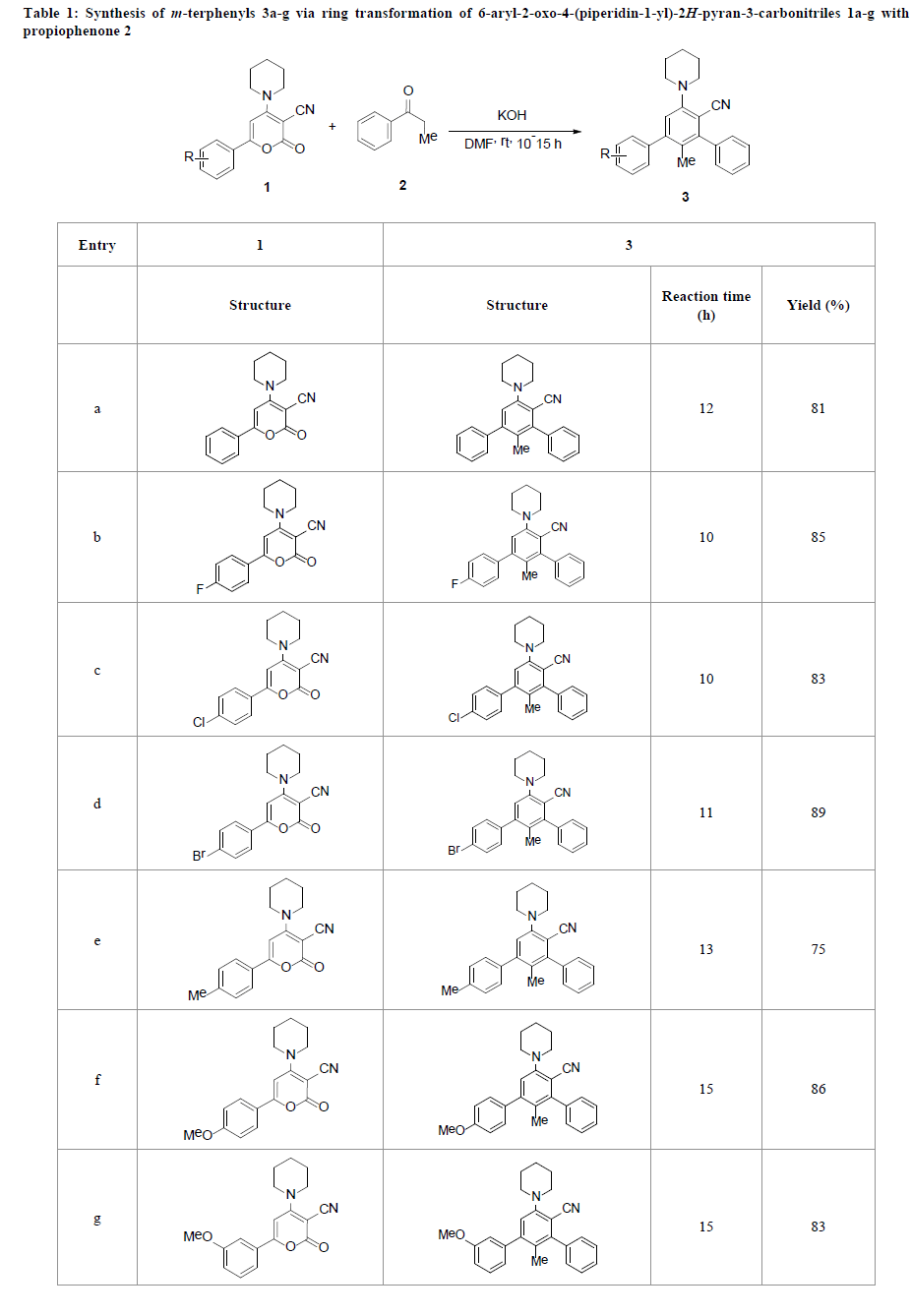
Our approach to prepare functionalized m-terphenyls 3a-g is based on the ring transformation of 6-aryl-2-oxo-4-(piperidin- 1-yl)-2H-pyran-3-carbonitriles 1a-g using propiophenone 2 as a carbanion source. The 2H-pyran-2-ones 1a–g precursors were synthesized by the reaction of ethyl 2-carbomethoxy-3,3-di(methylsulfanyl)-acrylate with substituted acetophenones under alkaline conditions followed by the reaction with cyclic amines [19]. Lactones, 1a–g has three electrophilic centres: C2, C4 and C6 in which the latter position is highly reactive towards nucleophiles due to the extended conjugation and the presence of the electronwithdrawing substitutent at position 3 of the pyran ring. In a typical process, stirring an equimolar mixture of 2H-pyran-2-ones 1a-g, propiophenone 2 and powdered KOH in DMF for 10-15 h at room temperature afforded functionalized m-terphenyls 3a-g in 75-89% yields (Table 1). Various electron-withdrawing and donating functionalities were successfully tolerated in the reaction. It was also observed that course of reaction was almost same with both electron-withdrawing and donating substituents on aromatic ring at C6 position of 2H-pyran-2one 3. All the synthesized compounds were characterized by the spectroscopic analysis.
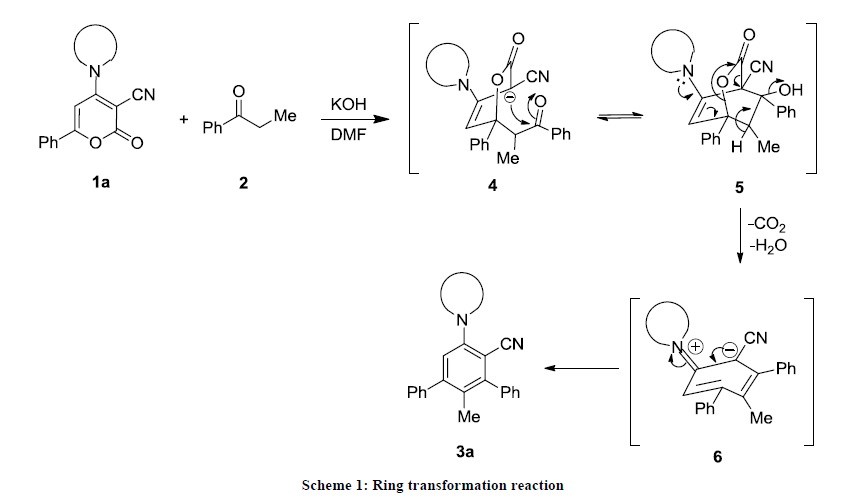
The possible mechanism of ring transformation reaction is depicted in Scheme 1. The transformation of 6-aryl-2H-pyran-2-ones 1 into m-terphenyls is possibly initiated by attack of the carbanion generated from 2 at position C6 of lactone 1a-g, followed by intramolecular cyclization involving the carbonyl functionality of 2 and C3 of the pyranone ring and elimination of carbon dioxide, followed by protonation and dehydration to yield 3a-g in good yields.
Conclusion
In summary, we have developed a new synthetic methodology for preparing functionalized meta-terphenyls through the carbanioninduced ring transformation of 2H-pyran-2-ones, in good yields. The methodology is very simple, economical and does not require any specialized organometallic reagents or catalysts. This methodology is a potentially useful alternative to conventional metalcatalyzed cross-coupling reactions.
Acknowledgements
Fateh V Singh and is thankful to Science and Engineering Research Board (SERB), New Delhi for providing financial support (Start- UP Research Grant: SB/FT/CS-068/2014). Authors are thankful to VIT University, Vellore Campus, for providing spectroscopic analysis.
References
[1] B.S. Yun, I.K. Lee, J.P. Kim, I.D. Yoo, J. Antibiot., 2000, 53, 114-122.
[2] H.J. Lee, I.K. Rhee, K.B. Lee, I.D. Yoo, K.S. Song, J. Antibiot., 2000, 53, 714-719.
[3] B.S. Yun, I.K. Lee, J.P. Kim, I.D. Yoo, J. Antibiot., 2000, 53, 711-713.
[4] L. Hu, J.M. Gao, J.K. Liu, Helv. Chim. Acta., 2001, 84, 3342-3349.
[5] S. Tsukamo, A.D. Macabalang, T. Abe, H. Hirota, T. Ohta, T. Tetrahedron., 2002, 58, 1103-1105.
[6] D.N. Quang, T. Hashimoto, Y. Hitaka, M. Tanaka, M. Nukada, I. Yamamoto, Y. Asakawa, Phytochemistry., 2003, 63, 919-924.
[7] T. Kamigauchi, R. Sakazaki, K. Nagashima, Y. Kawamura, Y. Yasuda, K. Matsushima, H. Tani, Y. Takahashi, K. Ishii, R.
Suzuki, K. Koizumi, H. Nakai, Y. Ikenishi, Y. Terui, J. Antibiot., 1998, 51, 445-450.
[8] K. Yasui, Y. Tamura, T. Nakatani, K. Kawada, M. Ohtani, J. Org. Chem., 1995, 60, 7567-7574.
[9] K. Yasui, Y. Tamura, T. Nakatani, I. Horibe, K. Kawada, K. Koizumi, R. Suzuki, M. Ohtani, J. Antibiot., 1996, 49, 173-180.
[10] K. Kawada, A. Arimura, T. Tsuri, M. Fuji, T. Komurasaki, S. Yonezawa, A. Kugimiya, N. Haga, S. Mitsumori, M. Inagaki, T.
Nakatani, Y. Tamura, S. Takechi, T. Taishi, J. Kishino, M. Ohtani, Angew. Chem. Int. Ed. Engl., 1998, 37, 973-975.
[11] S. Yonezawa, T. Komurasaki, K. Kawada, T. Tsuri, M. Fuji, A. Kugimiya, N. Haga, S. Mitsumori, M. Inagaki, T. Nakatani, Y.
Tamura, S. Takechi, T. Taishi, M. Ohtani, J. Org. Chem., 1998, 63, 5831-5837.
[12] R. Marchell, L.C Vining, J. Chem. Soc. Chem. Commun., 1973, 555-556.
[13] C. Takahashi, K. Yoshihira, S. Natori, M. Umeda, Chem. Pharm. Bull., 1976, 24, 613-620.
[14] H.G. Cutler, J.H. LeFiles, F.G. Crumley, R.H. Cox, J. Agric. Food Chem., 1978, 26, 632-635.
[15] I. Kurobane, L.C. Vining, A.G. McInnes, D.G. Smith, J. Antibiot., 1979, 32, 559-564
[16] F. Nakagawa, R. Enokita, A. Naito, Y. Iijima, M.J Yamazaki, J. Antibiot., 1984, 37, 6-9.
[17] A. Kobayashi, A. Takemoto, K. Koshimizu, K. Kawazu, Agric. Biol. Chem., 1985, 49, 867-868.
[18] C. Tringali, M. Piattelli, C. Geraci, G. Nicolosi, C. Rocco, Can. J. Chem., 1987, 65, 2369-2372.
[19] P. Stead, K. Affleck, P.J. Sidebottom, N.L. Taylor, C.S. Drake, M. Todd, A. Jowett, G. Webb, J. Antibiot., 1999, 52, 89-95.
[20] A.E. Sutton, J. Clardy, J. Tetrahedron. Lett., 2001, 42, 547-551.
[21] S. Chakraborty, C. Sengupta, K. Roy, Bioorg. Med. Chem. Lett., 2004, 14, 4665-4670.
[22] T.W. von Geldern, R.P. Brun, M. Kalmanovich, D. Wilcox, P.B. Jacobson, Synlett., 2004, 1446-1448.
[23] A.A. Greenfield, J.A. Butera, C.E. Caufield, Tetrahedron. Lett., 2003, 44, 2729-2732.
[24] F.V. Singh, A. Parihar, S. Chaurasia, A.B. Singh, S.P. Singh, A.K. Tamrakar, A.K. Srivastava, A. Goel, Bioorg. Med. Chem.
Lett., 2009, 19, 2158-2161.
[25] B.J. Groombridge, S.M. Goldup, I. Larrosa, I. Chem. Commun., 2015, 51, 3832-3834.
[26] H. Zhang, C. Wang, Z. Li, Z. Wang, Tetrahedron. Lett., 2015, 56, 5371-5376.
[27] J.M. Gadsby, C.B. McPake, C.B. Murray, G. Sandford, J. Flourine. Chem., 2016, 181, 51-55.
[28] Y. Sun, X. Li, H. Sun, Dal. Trans., 2014, 43, 9410-9413.
[29] Z. Xue, Y.Y. Qian, K.S. Chan, Tetrahedron. Lett., 2014, 55, 6180-6183.
[30] L. Gai, C.T. To, K.S. Chan, Tetrahedron. Lett., 2014, 55, 6373-6376.
[31] W. Liu, L. Xu, Tetrahedron. Lett., 2015, 71, 4974-4981.
[32] R.J.I. Tamargo, T.N. Poudel, Y.R. Lee, RSC. Adv., 2016, 6, 70311-70319.
[33] A. Goel, D. Verma, F.V. Singh, Tetrahedron. Lett., 2005, 46, 8487-8489.
[34] A. Goel, F.V. Singh, M. Dixit, D. Verma, R. Raghunandan, P.R. Maulik, Chem. Asian J., 2007, 2, 239-247.
[35] Y. Tominaga, A. Ushirogochi, Y. Matsuda, J. Heterocycl. Chem., 1987, 24, 1557-1567.



