Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 2
First-principles Study of Structural and Electronic Properties of Agn and Aun (n=3-4) Clusters Adsorbed on ZnO ÃÆà âÃâââ¬Â¦ Surface
Benkrima Yamina1*, Achouri Abderrahim1, Benmebrouk Lazhar1, Mohammedi Lazhar1, Bentouila Omar2 and Ouahab Abdelouahab31University of Ouargla, Faculty of Mathematics and Sciences of Matter, Laboratory Development of New and Renewable Energies in Arid and Saharan Areas, Ouargla-30000, Algeria
2Department of Matter Sciences, Faculty of Mathematics and Matter Sciences, LENREZA Laboratory, Optoelectronics Team, Kasdi Merbah, Ouargla University, Ouargla-30000, Algeria
3LPCMA, Thin Film Laboratory and Interfaces, University of Biskra-07000, Algeria
- *Corresponding Author:
- Benkrima Yamina
University of Ouargla, Faculty of Mathematics and Sciences of Matter
Laboratory Development of New and Renewable Energies in Arid and Saharan Areas
Ouargla-30000, Algeria
Abstract
Abstract
We perform first-principles calculations to investigate various surface structures of adsorbed Agn and Aun (n=3-4) clusters on a wurtzite ZnO (000͞1) surface. The results show that both Ag3 and Au3 clusters prefer to be adsorbed on the T4 (hcp-hollow) and Top sites of the surface while Ag4 and Au4 clusters prefer to be adsorbed on the T4 (hcp-hollow sites) of the ZnO (000͞1) surface. Agn/ZnO (000͞1) system is more stable than Aun/ZnO (000͞1) system due to a stronger binding energy. The calculated electron structure shows that the Agn and Aun clusters adsorbed on ZnO (000͞1) surfaces exhibit metallic characteristics. We calculated and discussed the functions of Agn and Aun clusters adsorbed on ZnO (000͞1) surfaces for the first time in the present work.
Keywords
Surface, Clusters, Adsorbed, Atomic structure, DOS, Catalysts
Introduction
Zinc Oxide ZnO is a direct wide band gap (ΔE=3.37 eV) semiconductor with a large excitation binding energy of 60 meV, which makes ZnO a relation versatile material applied in field effect transistors, solar cells, gas sensors, photodetectors and photocatalysts [1-5]. To expand more its application range, scientists apply various methods to modify the electronic structures of ZnO, for example depositing or doping various atoms into ZnO bulk materials both experimentally and theoretically.
Wurtzite ZnO is the most stable among all the structures of ZnO. Its surfaces have been extensively used as active catalysts and catalyst supports and, because of their opposite polarity (0001) and (000͞1) surfaces, have become the subject of intensive work in past years [6-9]. Noble metal/oxide systems have become an interesting field of investigation in the last years [10-12]. In these model systems, the interaction between the metal clusters and the oxides has a big role in the geometric and electronic structure of the resulting cluster/oxide interface, which in turn has a strong influence on the catalytic activity [13-15].
From late studies it is clear that ZnO supported Cu catalysts are important and become a fascinating subject of widespread investigations in recent years due to the practical applications. In deed surface properties become a little bit different from those of bulk copper [16]. From the theoretical side semi-empirical and accurate Density Function Theory (DFT) methods have been applied to study structures and electronic properties of atom or molecule deposition surfaces of ZnO [17-19].
There are theoretical and experimental studies for the surface structures and electronic properties of other noble metal clusters deposition on ZnO, such Agn and Aun [20]. For clarity noble metal adsorbed on the ZnO surface, we study in this paper the structures and electronic properties of Agn and Aun clusters deposition on wurtzite ZnO-O terminated (000͞1) surface by ab-initio calculations (DFT). In this work we concentrate on the size effect of supported metal clusters. The results allow us to make a direct comparison of these two metals clusters on ZnO (000͞1) surfaces. In this paper some useful information for future investigations on supported metal catalysts are provided.
Computational details
Our calculations are performed by using the first principles pseudo potential method based on DFT [21], the electronic exchange-correlation interactions were treated by the Generalized Gradient Approximation (GGA) with the (PBE) functional [22] as implemented in the SIESTA code [23]. The energy cutoff of the plane wave basis was chosen as 200 eV. The Brillouin Zones were sampled by very dense Monkhorst Pack K points grids of (6 × 6 × 1) for bulk ZnO, and (3 × 3 × 1) for ZnO (000͞1) surface and M/ZnO (000͞1) systems. Our self-consist-field (scf) calculation of total energy was carried out with a convergence criterion of 5 × 10-4 eV, and the maximum ionic movement tolerance within the cluster and atoms were set to 0.05 Å. The structure relaxation was done using the conjugated gradient method, when the atomic forces were smaller than 0.005 eV/Å.
For the bulk ZnO, the calculated lattice constants are a=b=3.284 Å and c=5.330 Å, which are in reasonable agreement with the corresponding experimental values of 3.25 Å and 5.207 Å [24]. We periodically repeated the slab supercell including four Zn-O double layers of ZnO. The slab possesses the same lattice spacing’s as those of the bulk in the x and y directions, but the atoms in z direction are relaxed.
The top two ZnO bilayers as well as the ad atom layer of the slab are allowed to be fully relaxed in the geometric optimization, while the two bottom ZnO bilayers are fixed. In addition, the dangling bonds on the bottom layer are saturated with pseudohydrogens in order to prevent unphysical charge transfer between the top and bottom slabs.
Results and Discussion
Structure and stability of the surface
The models of clean ZnO (000͞1) surface and M/ZnO (000͞1) system are simulated from the optimized bulk wurtzite ZnO unit cell. The optimized lattice constants by the (GGA) method are a=b=3.284 Å and c=5.33 Å, which are close to the experimental values of a=b=3.258 Å and c=5.22 Å [25]. We first optimized the clean wurtzite ZnO (000͞1) surface before investigating different structures of Agn or Aun adsorbed surfaces. The calculated results for the surface are listed in Table 1. The relaxed results of the distance between the uppermost four layers (R12, R23 and R34) are given by means of the difference from the un-relaxed ideal interlayer distance.
| Relaxation interlayer | Difference between the relaxed distance and the initial distance | |
|---|---|---|
| This work | Ref [26] | |
| R12 (Å) | -0.309 | –0.301 |
| R23 (Å) | 0.082 | 0.112 |
| R34 (Å) | -0.104 | –0.083 |
Table 1: Relaxation of the distance between the uppermost four layers.
The Zn-O monolayer spacing’s for the first, second, and third bilayers are -0.309 Å, 0.082 Å, -0.104 Å respectively, in good, agreement with the theoretical results of -0.301 Å, 0.112 Å and -0.083 Å [26]. We can see that polar surface ZnO (000͞1) tend to relax inward, because surface relaxation is mainly affected by Coulomb force and hybrid quantum effect. The energy of the surface relaxation is greater than the energy of the unrelaxation surface, so the surface relaxation of ZnO (000͞1) is most stable. For the study of Agn and Aun (n=3-4) clusters adsorbed on the ZnO (000͞1) surfaces, a slab model with 3 × 3 surface periodic supercell consisted of four Zn-O double layers (Figure 1).
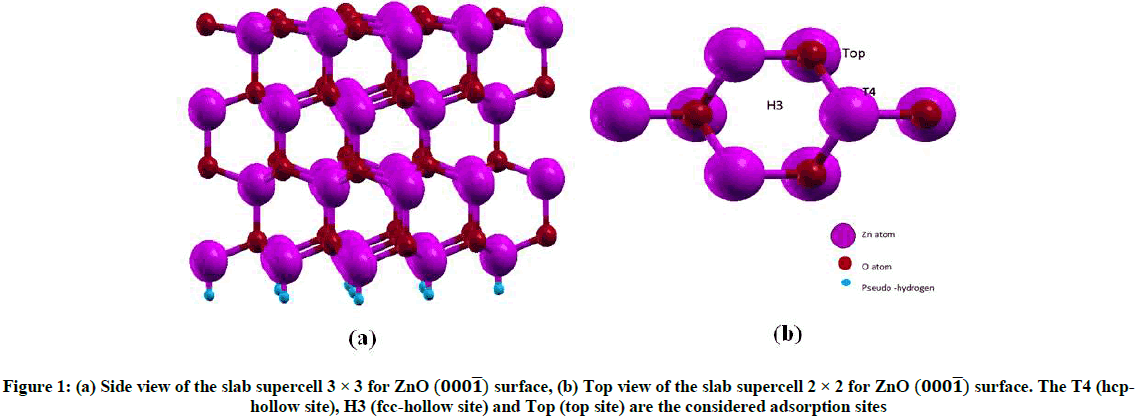
Figure 1: (a) Side view of the slab supercell 3 × 3 for ZnO (000͞1) surface, (b) Top view of the slab supercell 2 × 2 for ZnO (000͞1) surface. The T4 (hcp-hollow site), H3 (fcc-hollow site) and Top (top site) are the considered adsorption sites
To further investigate the influence of the adsorption sites to the stability of Agn and Aun (n=3-4) clusters on the ZnO (000͞1) system, four relative stable structures have been selected. The optimized structures are shown in Figure 2. Clusters used in the adsorption process in this study were calculated using the simulated annealing process’ method, where heating to a high temperature around 1000 K was performed in 1000 steps. Then, they were equilibrated at this temperature in another 1000 steps, and slowly cooled down to 0 K in 500 steps. Finally, the clusters were allowed to relax at 0 K. We find that both Ag3 and Au3 clusters prefer to be adsorbed on the T4 (hcp-hollow sites) and the Top sites of the ZnO (000͞1) surface while Ag4 and Au4 clusters prefer to be adsorbed on T4 (hcp-hollow sites) of the ZnO (000͞1) surface.
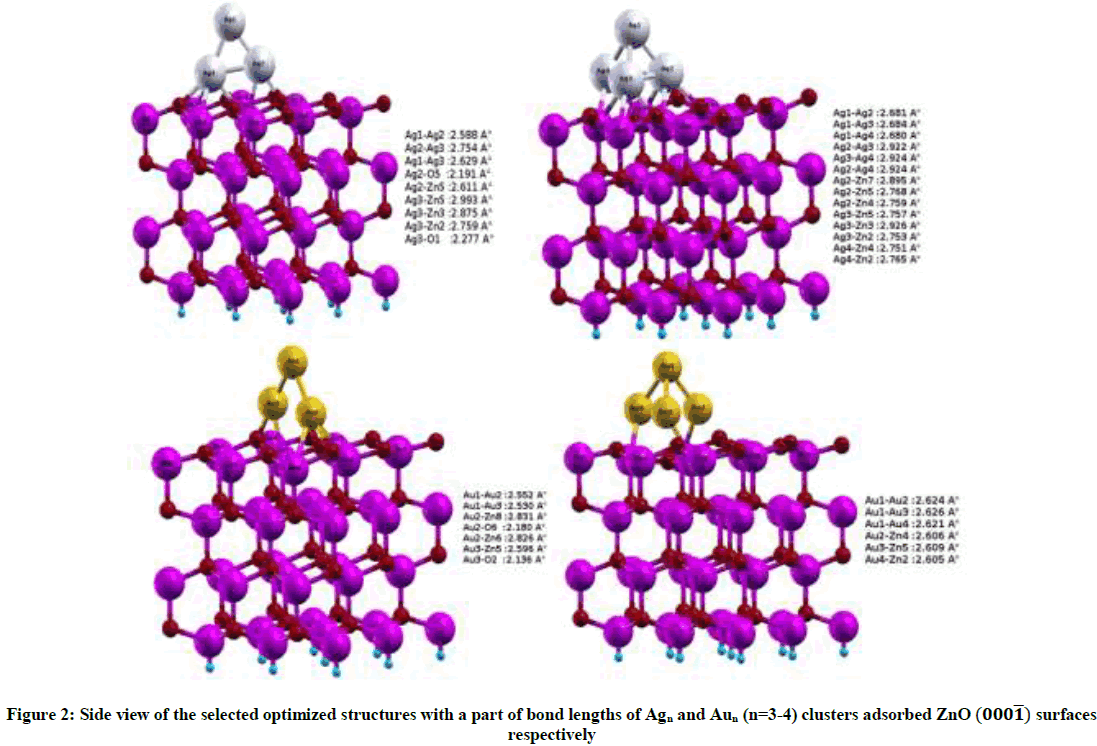
Figure 2: Side view of the selected optimized structures with a part of bond lengths of Agn and Aun (n=3-4) clusters adsorbed ZnO (000͞1) surfaces respectively
The adsorbed Agn or Aun clusters not only bond with surface O atoms, but also bond with each other. The Ag-Ag or Au-Au bond that leads has the lowest energy and is the most stable adsorption structure. So in the process of adsorption on the ZnO surface Agn and Aun clusters are preferred to connect with Zn atoms with very few bonds with O atoms.
The calculated energies for different interaction modes of Agn and Aun clusters with the surface are listed in Table 2. In Table 2 one can see that the adsorption of Ag3 on the surface ZnO (000͞1) is energetically more favorable than the cluster Au3. It’s the same observation for Ag4 and Au4.
| Clusters | Energies (eV) |
|---|---|
| Ag3 | -72947.3155 |
| Ag4 | -73931.7662 |
| Au3 | -72690.0802 |
| Au4 | -73587.9086 |
Table 2: The calculated energies of M/ZnO (000͞1) (M = Agn and Aun clusters) surfaces.
Binding energy
The mean interaction (binding) energy per atom of the cluster to the surface is calculated including those atoms which are not in direct contact with the surface for two reasons: (1) The non-bonded atoms interact with the bonded ones and can be considered as interacting indirectly with the surface, and (2) This is the only way to compare with the interaction energy for clusters of different size. The binding energy per atom, EB can be estimated by the following Equation:
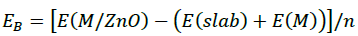 (1)
(1)
Where, n is the number of Ag or Au atoms, E(M/ZnO) is the total energie of Agn or Aun adsorbed on the surface, and E(M) are energies of Agn or Aun clusters respectively. The more negative value of EB, he more stable the M/ZnO (000͞1) system. The calculated binding energies of Agn and Aun/ZnO (000͞1) system are listed in Table 3.
| System | Binding energies (eV) |
|---|---|
| Ag3/ZnO(000͞1) | -2.255 |
| Ag4/ZnO(000͞1) | -2.083 |
| Au3/ZnO(000͞1) | -1.997 |
| Au4/ZnO(000͞1) | -1.753 |
Table 3: The calculated binding energies of Agn and Aun on ZnO(000͞1) surfaces.
The tabulated binding energies show clearly that, for the polar surface ZnO (000͞1) , the binding energy of the Ag3/ZnO system is greater than for the Ag4/ZnO system, and Au4/ZnO system is less active in binding than the cluster Au3 adsorbed of the ZnO (000͞1) surface. Comparing the binding energies of Agn/ZnO and Aun/ZnO, it is found that the binding of Ag3/ZnO is stronger with the binding energy greater by -0.26 eV than that in Au3/ZnO, and the same result of Ag4/ZnO is stronger with the binding energy greater by -0.33 eV than that in Au4/ZnO system. M/ZnO (000͞1) system with adsorbed Agn clusters is more stable than with adsorbed Aun clusters. This reveals that the chemical activities of Agn clusters adsorbed on ZnO (000͞1) are more than Aun clusters adsorbed on the same surface.
Electronic properties
We calculate the electron structure for the bulk wurtzite ZnO after the structure optimization by using the same method. Our calculated results show that the band gap of the bulk is 0.45 eV, in good agreement with the theoretical results [27], which is lower than the experimental value. This is regarded as a spread problem of DFT calculations (Figure 3). The bulk ZnO calculation imply semiconductor characteristics as it is found in the literature [27].
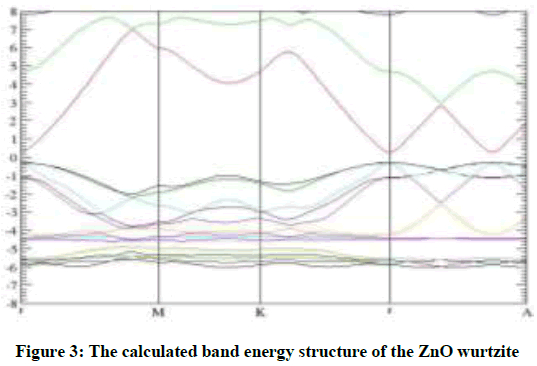
Figure 3: The calculated band energy structure of the ZnO wurtzite.
Density of states
As we know, Ag and Au are inactive in bulk, but they turn into more active catalysts for ZnO nanostructures when small clusters of Agn or Aun are deposited on the ZnO (000͞1) surface. To make clear the way for this change in properties, we performed a comparative study of the electronic structure of the metal/oxide interface and the free surface. To find the electronic properties of Agn and Aun clusters adsorbed on ZnO (000͞1) surfaces, the density of states DOS of the system with a 3 × 3 surface cell for Agn and Aun clusters adsorbed on ZnO (000͞1) surfaces are presented in Figure 4.
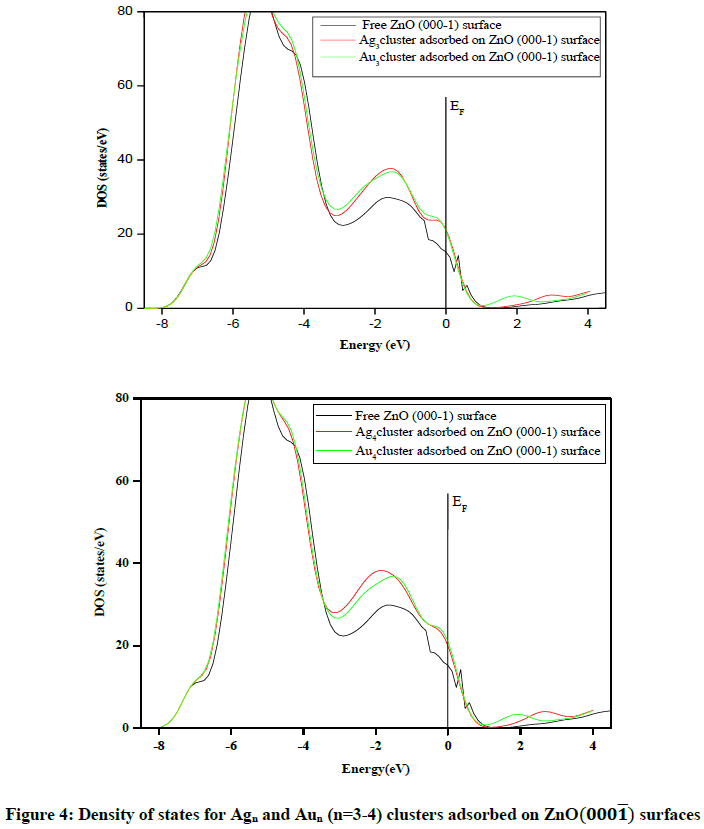
Figure 4: Density of states for Agn and Aun (n=3-4) clusters adsorbed on ZnO (000͞1) surfaces.
The energy Zero (marked by a vertical line) is aligned at the Fermi level. The M/ ZnO (000͞1) system is quite different from the surface ZnO (000͞1). The density of states gives important information about the ‘available’ charge (electron or fraction of it) for a given energy. For example, chemical activity of a metal/oxide is proportional to the electron density near the Fermi level. The higher the density in this region, the higher the chemical activity. In Figure 4, we present the plot of the DOS variation for the M/ZnO (000͞1) system. As we can see, the highest DOS near Fermi level is present for Ag3, Ag4, Au3 and Au4, while the free ZnO surface has little DOS in this region.
Two interesting features are observed immediately from Figure 4; First, the electronic structures of Agn and Aun clusters adsorbed by ZnO (000͞1) surfaces are the same. Second, the DOS shows that these surfaces exhibit metallic characteristics. This property is similar to the results obtained from adsorption of Ag and Au atoms on wurtzite ZnO (0001) surface [28]. Through this analysis, we can predict that Agn clusters can be the most chemically active with a ‘selective’ activation energy due to the energy distance variation from the Fermi level which is a very important property for catalysis. This notable energy of EF for the M/ZnO compared to that of the free ZnO surface contributes in the enhanced catalytic activity of M/ZnO system.
Conclusion
In this paper, we investigate the structures, stabilities and electronic properties of Agn and Aun clusters adsorbed on ZnO (000͞1) surfaces are systematically investigated. We find that both Ag4 and Au4 clusters prefer to be adsorbed on T4 (hcp-hollow sites) of the ZnO (000͞1) surface, and Ag3 and Au3 clusters prefer to be adsorbed on the T4 (hcp-hollow) and the Top sites of the ZnO (000͞1) surface. In DOS analysis we find that the Agn and Aun clusters adsorbed on ZnO (000͞1) surfaces exhibit metallic characteristics. These conclusions were obtained using firstprinciples density functional theory calculations. Finally, the work function of Agn and Aun clusters adsorbed on ZnO (000͞1) surfaces are calculated and discussed for the first time in the paper. We wish the present work provided useful information about noble metal clusters adsorbed on ZnO surfaces.
References
- J. Goldberger, D.J. Sirbuly, M. Law, P. Yang, J. Phys. Chem B., 2005, 109, 9.
- K. Keis, L. Vayssieres, S.E. Lindquist, A. Hagfeldt, Nanostruct. Mater., 1999, 12, 487.
- Q. Wan, Q.H. Li, Y.J. Chen, T.H. Wang, X.L. He, J.P. Li, C.L. Lin, Appl. Phys. Lett., 2004, 84, 3654.
- H. Kind, H.Q. Yan, B. Messer, M. Law, P.D. Yang, Adv. Mater. 2002, 14, 158.
- S. Sakthivel, B. Neppolian, M.V. Shankar, B. Arabindoo, M. Palanichamy, V. Murugesan, Sol. Energy Mater. Sol. Cells., 2003, 77, 65.
- J. Hu, W.P. Guo, X.R. Shi, B.R. Li, J.G. Wang, J. Phys. Chem. C., 2009, 113, 7227.
- V. Ney, S. Ye, T. Kammermeier, A. Ney, H. Zhou, J. Fallert, H. Kalt, F.Y. Lo, A. Melnikov, A.D. Wieck, J. Appl. Phys., 2008, 104, 083904.
- P. Lazcano, M. Batzill, U. Diebold, P. Haberle, Phys. Rev. B., 2008, 77, 035435.
- N.S. Phala, G. Klatt, E. van Steen, S.A. French, A.A. Sokol, C.R.A. Catlow, Chem. Chem. Phys., 2005, 7, 2440.
- C.T. Campbell, Surf. Sci. Rep., 1997, 27, 1.
- J. Yoshihara, S.C. Parker, C.T. Campbell, Surf. Sci., 1999, 439, 153.
- H.J. Freund, Surf. Sci., 2002, 500, 271.
- M. Haruta, Chem. Rec., 2003, 3, 75.
- H. Nakajima, T. Mori, M. Watanabe, J. Appl. Phys., 2004, 96, 925.
- V. Subramanian, E.E. Wolf, P.V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943.
- J. Yoshihara, J.M. Campbell, C.T. Cambell, Surf. Sci., 1998, 406, 235.
- J.B.L. Martins, E. Longo, C.A. Taft, Int. J. Quant. Chem., 1998, 70, 367.
- X.Q. Dai, H.J. Yan, J.L. Wang, Y.M. Liu, Z.X. Yang, M.H. Xie, J. Phys. Condens. Matter., 2008, 20, 095002.
- P. Persson, L. Ojamäe, Periodic, Chem. Phys. Lett., 2000, 321, 302.
- E. Gyorgy, J. Santiso, A. Figueras, A. Giannoudakos, M. Kompitsas, I.N. Mihailescu, C. Ducu, Appl. Surf. Sci., 2006, 252, 4429.
- D. Vanderbilt, Phys. Rev. B., 1990, 41, 7892.
- J.P. Perdew, S.M. Burke, H. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865.
- D. Sánchez- Portal, P. Ordejón, E. Artacho, J.M. Soler, Int. J. Quant. Chem., 1997, 65, 453.
- C.H. Bates, W.B. White, R. Roy, Science., 1962, 137, 993.
- F. Decremps, F. Datchi, A.M. Saitta, A. Polian, S. Pascarelli, A. Di. Cicco, J.P. Itie, F. Baudelet, Phys. Rev. B., 2003, 68, 104101.
- Z. Yufei, G. Zhiyou, G. Xiaoqi, C. Dongxing, D. Yunxiao, Z. Hongtao, J. Semicond., 2010, 31, 082001.
- B Haq, A. Afaq, R. Ahmed, S. Naseem, Int. J. Mod. Phys., 2012, 23, 1250043.
- Z. Yang, S.J. Xiong, Surf. Sci., 2011, 605, 40.



