Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 12
Formulation, In vitro and In vivo Analysis of Cyclodextrin Complexed Albendazole Composites for Enhanced Solubility
Anjana MN*
Department of Pharmaceutics, Pushpagiri College of Pharmacy, Medicity Campus, Peruthuruthi P.O., Thiruvalla, Kerala, India
- *Corresponding Author:
- Anjana MN
Department of Pharmaceutics
Pushpagiri College of Pharmacy
Medicity Campus, Peruthuruthi P.O., Thiruvalla, Kerala, India
Abstract
Albendazole is a low water soluble benzimidazole carbonate drug, extensively used against instestinal parasites due to its broad spectrum activity, good tolerance and low cost. The drug has a disadvantage of poor bioavailability due to very low solubility in water. The main objective of the study was to increase the solubility and dissolution rate by complexing with cyclodextrins by kneading method and to compare the solubility between hydroxypropyl beta cyclodextrin and beta cyclodextrin. Solid state characterization was done by Fourier Transform Infrared (FTIR) and thermo gravimetric analysis. Increased solubility was obtained when more substituted cyclodextrins were used instead of non-substituted cyclodextrins. High oral dose of albendazole is required for treating systemic helminthiasis which can lead to liver impairment, so the main aim of the study was to analyze liver enzymes using albino wistar rats. For enzyme determination, blood samples were collected from each rat after 48 h of drug administration via retro-orbital puncture method and allowed to clot. From the studies it was found that elevation in liver enzymes is less in case of ABZ-HPβCD complex suspensions than the other formulations. Drug release studies and kinetic profiles help to find out the release of drug from cyclodextrins and it was found out that ABZ-HPβCD suspension having better release profile than the ABZ-βCD.
Keywords
Liver enzymes, Cyclodextrins, Solubility, Albino wistar rats, Liver toxicity, Albendazole.
Introduction
Solubility is one of the important parameters to achieve desired concentration of drug in systemic circulation for achieving required pharmacological response. Poor water soluble drugs often require high doses in order to reach the therapeutic plasma concentration after oral administration. Low solubility is the major problem encountered with formulation development of new chemical entities as well as generic development. The poor solubility and low dissolution rate of poorly soluble drugs in the gastro intestinal fluids often causes insufficient bioavailability. Albendazole is a wide spectrum lipophilic anthelmintic drug belongs to biopharmaceutical classification system type II: low solubility and bioavailability and high permeability. ABZ belongs to a group of benzimidazole derivative with broad spectrum of activity against human and animal helminthe parasites such as nematodes, hydatodes and metacestodes. But it is well known that the drug was poorly or erractically absorbed from gastro intestinal tract, but is still drug of choice for the treatment of echinococcosis, hydrates cysts, and neurocycticercosis [1]. ABZ the drug candidate in the current studies is a poor water soluble drug, known to demonstrate dissolution limited absorption. ABZ when administered orally is quickly biotransformed into its active metabolite albendazole-sulphoxide, which is then oxidized to the inactive form of albendazole-sulphone. Because of their affinity for the parasite b-tubulin, both albendazole and albendazole-sulphoxide shows anthelmintic activity. Futher more lack of water solubility reduces flexibility for albendazole formulations and administration. Therefore lots of efforts have made to improve the aqueous solubility of albendazole. One of the possible ways to overcome this problem is to alter the physical properties of the drug by forming complexes with cyclodextrins [2]. Commercially ABZ is available in tablet and suspension. The choice of suspension instead of tablet and capsule is frequently governed by patience acceptance. Major disadvantage in the treatment of systemic helminthiasis was the poor bioavailability of albendazole. In some cases high doses therapy by the oral route lead to adverse reaction such as gastro intestinal disturbances and liver impairement. Damage or injury to liver cause elevations to the liver enzyme levels and elevations in serum enzyme levels are taken as the relevant indicators of liver toxicity. Administration of albendazole can cause elevation in liver enzymes, so enzyme analysis was performed to find out any elevation in alanine amino transferase, aspartate aminotransferase, and alkaline phosphatase [3]. The main objective was to improve the solubility of albendazole by complexing with hydroxyl propyl beta cyclodextrin and beta cyclodextrins and to compare the effect of both cyclodextrins on albendazole. Cyclodextrins are structurally related natural products they belong to the family of cyclic oligosaccharides with a hydrophyllic outer surface and a lipophilic central cavity, and are formed by the bacterial digestion of celloulose [4]. The binary system containing Albendazle and cyclodextrins is prepared by kneading method (ABZ/HPβCD and ABZ/ βCD), and solubility comparison is done between pure albendazole and the kneaded mixtures. The objective of present study was to evaluate the metabolic changes that could be encountered in response to albendazole administration. One of the side effects of albendazole, on frequent administration can lead increase in liver enzymes so objective of the study was to find out the effect of albendazole and kneaded mixtures on liver enzymes. Currently there are approximately 30 different pharmaceutical products worldwide containing drug/cyclodextrin complexes in market, examples are itraconazole oral/iv solution, omeprazole oral tablet and chloramphenical eye drops.
Materials and Methods
Albendazole is obtained as gift sample from Cipla Ltd. Mumbai, hydroxyl propyl beta cyclodextrin and beta cyclodextrin from Yarrowchem Products, Mumbai. All other chemicals used in this study were of analytical grade. Before proceeding to the formulation part, pre formulation studies are first conducted mainly to investigate the physical and chemical properties of drug substance alone and when combined with excipients.
Determination of λmax [5]
An absorption maximum of Albendazole was determined using 0.1 N HCl (pH 1.2). Solution ranging from 10-100 μg/ml were scanned from 200-400 nm using UV spectrophotometer.
Melting point determination [6,7]
Melting range was determined by capillary fusion method. A capillary was taken and the open end of the capillary tube was pushed into a small heap of ABZ so that a small plug of the powder was collected in the open end. The tube was tapped gently, so that the collected material was allowed to settle down. Then the capillary tube was placed in the melting point determination apparatus. The temperature at which Albendazole starts to melt was noted.
Formulation and evalution of albendazole composites
Physiochemical characterization
Determination of partition coefficient [8]: Partition coefficient of Albendazole in n-octanol-water was determined. Equal volume of water and n-octanol (15 ml) were taken in separating funnel. To this known amount of Albendazole was added. The funnel was equilibrated for 2 h at constant temperature with intermittent shaking at regular intervals. Then the aqueous and n-octanol layers were separated. The concentration of solute in the aqueous layer was determined by UV spectroscopy at 229 nm. The n-octanol-water partition coefficient of the drug was obtained using the following equation.
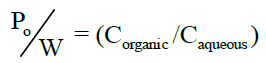
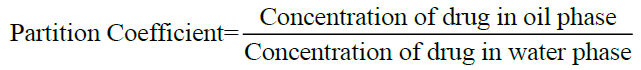
Concentration in organic phase can be calculated by using the equation,
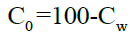
Determination of solubility [9]: Solubility of the drug was determined in different solvents such as strong acids like hydrochloric acid, sulphuric acid, strong bases like sodium hydroxide and in methanol, chloroform, ethyl acetate, glacial acetic acid and in water.
Drug-excipient compatibility study [10]: Excipients were important ingredients of almost all pharmaceutical dosage form. The successful formulation of a stable and effective solid dosage form depends on the selection of excipients, so it is necessary to know the inherent stability of the drug substance and possibility of interaction with excipients. The physical compatibility studies were coupled with the stability studies at higher temperature and humidity conditions. The physical mixtures were sealed into and stored in humidity chamber for 30 days at 40°C temperature and 75% relative humidity conditions. The initial state of the mixtures was noted and a further evaluation for the possible occurrence of any interaction was performed after 15th and 30th day.
Preparation of binary mixture of ABZ/HPβCD and ABZ/ βCD
In this study binary mixture of Albendazole /HPβCD and Albendazole/β CD is prepared by kneading method.
Complexation of ABZ/ HPβCD and ABZ/βCD by kneading technique
Albendazole/hydroxyl propyl β cyclodextrin (1: 1.5) and Albendazole βCD (1: 1.5) were prepared by kneading method. In this method hydroxyl propyl β cyclodextrin is taken in a glass mortar, little water was added mixed to obtain homogenous paste. The drug was added slowly while grinding. The mixture was ground for 1 h and during this process approximate quantity of water was added to maintain suitable consistency. The paste was dried in hot air oven at 40°C for 48 h. The dried complex was further passed through 60#mesh and packed in closed container; same procedure is used for the preparation of ABZ/βCD.
Preparation of oral suspensions: Suspensions containing 40 mg/ml were prepared as per the formula given in Table 1, in about 100 ml of purified water.
| Ingredients | βCD (1: 1.5) g | HPβCD (1: 1.5) g |
|---|---|---|
| Albendazole | 4 | 4 |
| HP β CD | 4 | 6 |
| Sodium CMC | 1 | 1 |
| Tween 80 | 1 | 1 |
| Sodium benzoate | 0.8 | 0.8 |
| Sucrose | 30 | 30 |
| Pineapple flavour | 0.3 | 0.3 |
| Tartarzine colour | 0.0001 | 0.0001 |
| Purified Water | q.s | q.s |
| Total (ml) | 100 ml | 100 ml |
Table 1: Composition of HPβCD-ABZ and βCD-ABZ Suspension
Comparison of solubility [8]: Solubility studies were carried out in distilled water.100 mg of pure drug, kneaded mixture (ABZ-HPβCD and ABZ-βCD) were placed in 50 ml of water and kept in an orbital shaker for 24 h at 25°C. The solutions were filtered and the concentration was determined by UV-spectrophotometer at 229 nm. The results were extrapolated to determine the percent w/w of albendazole.
In vitro dissolution studies [11]: USP XXIII dissolution test apparatus-II (Veego digital tablet dissolution test apparatus, model VDA-8D) was used for dissolution testing of both βCD and HPβCD complexed Albendazole suspension in these studies. A digitally controlled water circulator/heater was used to maintain the temperature of the media at 37°C ± 0.5°C and the dissolution medium was 0.1 N HCL (pH 1.2), and the volume of dissolution medium was 900 ml. The dissolution test was conducted at an agitation speed of 50 rpm. Samples (3 ml) were withdrawn and replaced with 3 ml of fresh medium to maintain a constant volume. Samples were collected at predetermined time intervals after 5, 20, 40, 70, 90, 110, and 120 min and quantified using UV spectrophotometer at 229 nm for determination of the albendazole content using the calibration curve. Each formulation was tested in triplicate and the mean values are calculated.
Kinetic studies [12]: Graph Pad prism software Version was used to fit the dissolution data generated following testing of all formulation. The release data obtained was fitted into various kinetic models like Zero order, First order, Korsmeyer-Peppas model and Higuchi Model. The best fit solution was identified by evaluating the coefficient of determination (R2) and the highest R2 value indicate the best fit. Following are the mathematical equations for the models used to describe the dissolution curves for formulations.
Zero order kinetics: A zero order release can be predicted by using the equation:
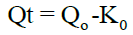
First order kinetics: The first order describes the release from system where the release rate is concentration dependent. It can be described by following equation:
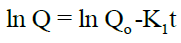
Higuchi Model: The drug release can be predicted by the following equation:
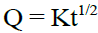
Korsmeyer Peppas model
To evaluate the mechanism of drug release, the in vitro release data was plotted in Korsmeyer equation as log cumulative percentage of drug release vs log time and the exponent n was calculated through the slope of straight line.
Korsmeyer equation is as follows:
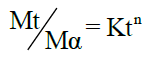
Where, n is an exponent which indicates the mechanism of drug release. The plots were drawn using excel sheet and the regression equations were obtained for each plot. The linearity of the plots was obtained from the value of regression coefficient (R2). The model with the highest linearity (R-value approaches unity) was chosen as the best-fit kinetic model.
Zeta potential determination
Zeta potential is a measure of the magnitude of the electrostatic repulsion/attraction between particles, and is one of the fundamental parameters known to affect stability. The zeta potential of the HPβCD/ABZ and β CD/ABZ was determined by Zetasizer.
Solid state characterization studies
The FTIR spectrum was obtained on an IR spectrophotometer from 3500 to 600 cm-1. The FTIR spectrum obtained from pure drug of Albendazole I.P was compared with the FTIR of the standard drug spectrum.
DTA thermogram of albendazole [2,14]
The samples were analysed by Thermo gravimetric analyser (TG/DTA-6200, EXSTAR). The samples (5 mg each) were placed in aluminium crucible. The studies were performed by recording the heat flow rate from 23º-450º; at a rate of 10°C/min. Aluminium was used as a standard reference material to calibrate the temperature and energy scales of DTA instrument. The peak temperatures were determined after calibration with standard.
Compatibility of Albendazole with the individual excipient and physical mixture of main formulation was established by Infrared Absorption Spectral Analysis (FTIR). Any change in the chemical composition after combining with the excipients were investigated with IR spectral analysis.
In vivo studies
The protocol for the animal study prescribed in proforma B was submitted to the Institutional Animal Ethics Committee (IAEC) of the Amrita Institute of Medical Sciences, Kochi. The protocol was approved by the IAEC.
Liver enzyme analysis using Wistar albino rats [3,15]
Wistar albino male rats weighing 280 ± 20 g were used for this study. Albino rats were divided into four groups and were fasted overnight. First group of animal received water as control, second group animal received β CD/ABZ orally at a dose of 40 mg/kg body weight third animal received HPβCD/ABZ orally at a dose of 40 mg/kg/body weight and fourth group received marketed formulation. Blood samples were obtained from control group as well as treated group after 48 h of drug administration. For enzyme determination, blood samples were collected from each rat after 48 h of drug administration via retro-orbital puncture method and allowed to clot. The serum was rapidly separated by centrifuging the clotted blood at 3000 rpm for 10 min and processed for determination in to clean and dry tubes. Sera were stored at -20ºC until assayed for the biochemical parameters. Biochemical analyses were carried out to determine the activities of liver enzymes such Alanine amino transferase, Aspartate aminotransferase, and alkaline phosphatase using diagnostic kits.
Results and Discussions
λmax of the drug
From the figure it can been seen that the spectrum of Albendazole has a maximum absorbance at 229 nm in 0.1 N HCL, it obeys Beer Lambert law and the absorbance value was in accordance with the official standard, and it shown in Figure 1.
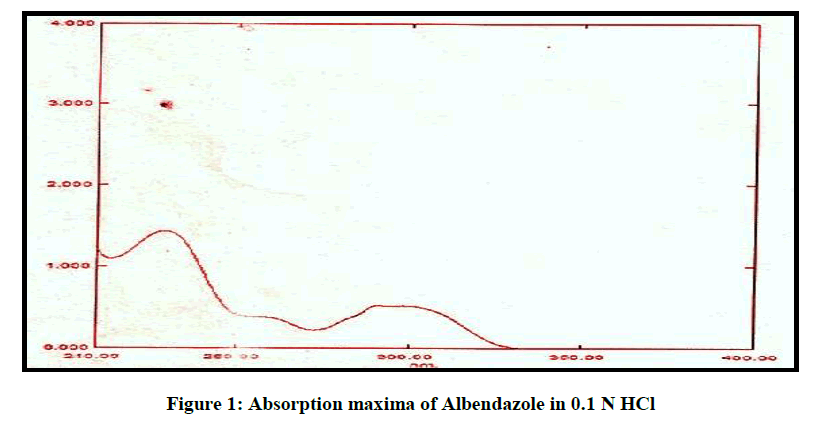
Figure 1: Absorption maxima of Albendazole in 0.1 N HCl
Melting point determination
The melting point of Albendazole were determined by open capillary method and it was found to be 205ºC, this value is same as that of literature citation of 204°C-209ºC.
Solubility of the drug
The solubility of the pure drug was compared with reference sample and it tabulated in Table 2. It reveals that the pure drug is insoluble in water and freely soluble in concentrated acids, concentrated bases, glacial acetic acid, formic acid, dimethylformamide. The + sign indicates that drug is soluble in that solvent and – sign indicates drug is insoluble.
| Solvent | Solubility | |
|---|---|---|
| Reference | Observation | |
| Water | - | - |
| 0.1 NHCL | + | + |
| Concentrated HCL | + | + |
| Formic acid | + | + |
| Glacial acetic acid | + | + |
| Dimethyl formamide | + | + |
| Concentrated NaOH | + | + |
Table 2: Comparison of solubility profile of pure drug with reference
Partition coefficient of the drug
The partition coefficient of the drug was found to be 3.22. This indicates that Albendazole is practically insoluble in water and having high lipid solubility.
Physical drug excipient compatibility
The physical mixtures of drug were prepared in 1: 1 ratio and from the drug excipients compatibility study, it was observed that there were no incompatibilities or interaction between drug and excipients and is shown in Table 3. The compatibility of drugs with excipients was studied by FTIR.
| No. | Drug + Excipients | Description (Observations) | ||
|---|---|---|---|---|
| Initial | 2nd Week | 3rd Week | ||
| 1 | Pure Albendazole | White Colour | No Colour Change | No Colour Change |
| 2 | Albendazole + HPβCD | White Colour | No Colour Change | No Colour Change |
| 3 | Albendazole + βCD | White Colour | No Colour Change | No Colour Change |
Table 3: Drug excipient compatibility
IR spectral analysis of ABZ
The FTIR spectrum of the drug is given in Figure 2, it was found in accordance with the reference standard. The principle peaks of the IR spectra matched the reference listed peaks of Albendazole and principle peaks are given in Table 4. Kneaded mixture of ABZ showed superimposed spectra of ABZ and hydroxy propyl beta cyclodextrin which proves the compatibility of excipients with the albendazole, given in Figure 3.
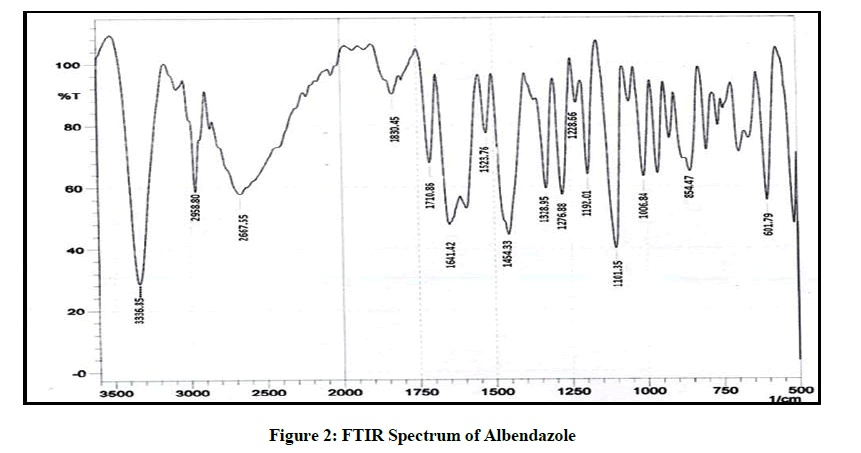
Figure 2: FTIR Spectrum of Albendazole
| No. | Frequencies (cm-1) | Assignment |
|---|---|---|
| 1 | 3336.85 | N-H Streching of amine |
| 2 | 2958.8 | Strenching of alkane |
| 3 | 1710 | C=O Strenching of ketone |
| 5 | 1524 | Bending Vibration |
Table 4: Principal Peaks of IR Spectrum of ABZ
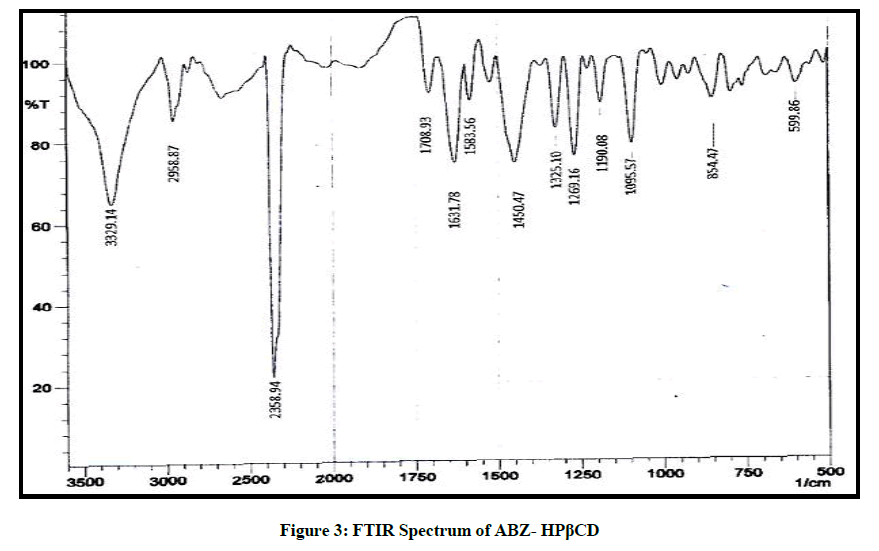
Figure 3: FTIR Spectrum of ABZ- HPβCD
DTA of Albendazole
DTA of Albendazole exhibits an endothermic peak at 203ºC (Figures 4-7). The sharp endothermic peak at this region is due to the melting of crystalline Albendazole. From DSC it was confirmed that the drug is crystalline in nature and has a melting point 203ºC i.e., with an associated weight loss, which comprised two processes as seen by lack of symmetry within the peak and uniformity in weight loss and followed by endothermic peak there is an exothermic peak at 348ºC may be due to the 2nd weight loss. The DTA curve of HPβCD shows its thermal decomposition which begins around 280ºC and peaks are exothermic in nature. In the ABZ: βCD obtained by kneading mixing, the DTA technique shows the endothermic peak corresponding to the melting of ABZ. The presence of this peak indicates that a true inclusion complex has not been achieved. In the ABZ: HPβCD system obtained by kneading method, the endothermic peak corresponding to the melting of ABZ is not present. A new peak appears at temperature between 320ºC and 380ºC that can be attributed to melting of true inclusion complexes.
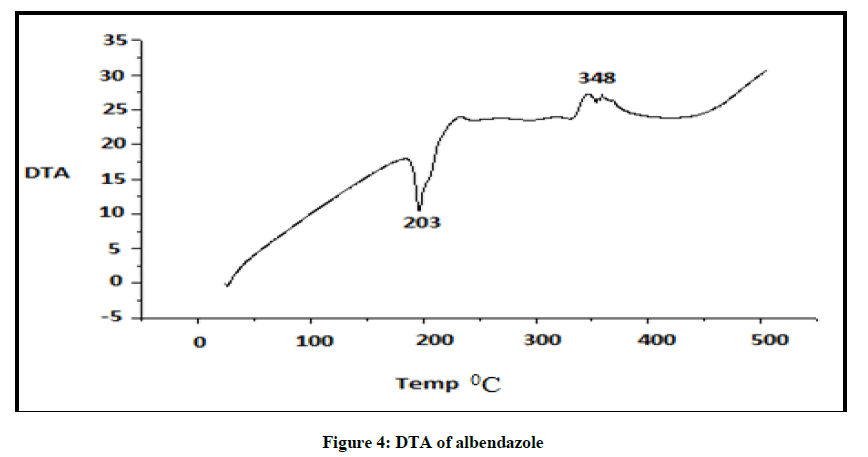
Figure 4: DTA of albendazole
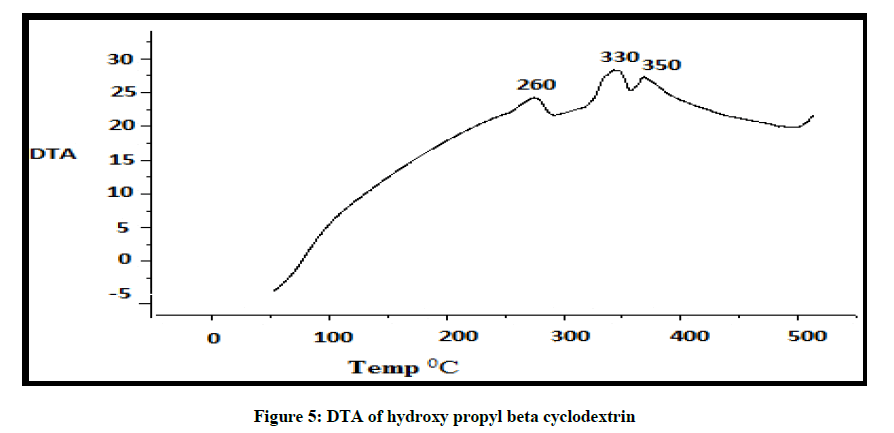
Figure 5: DTA of hydroxy propyl beta cyclodextrin
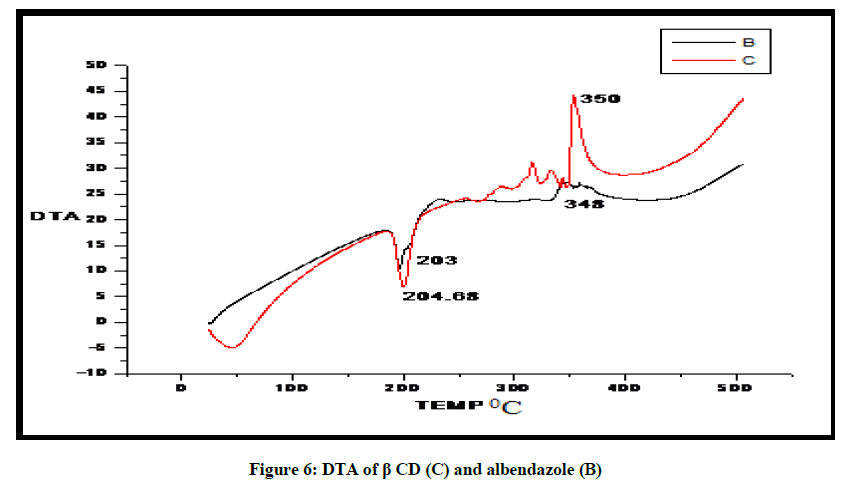
Figure 6: DTA of β CD (C) and albendazole (B)
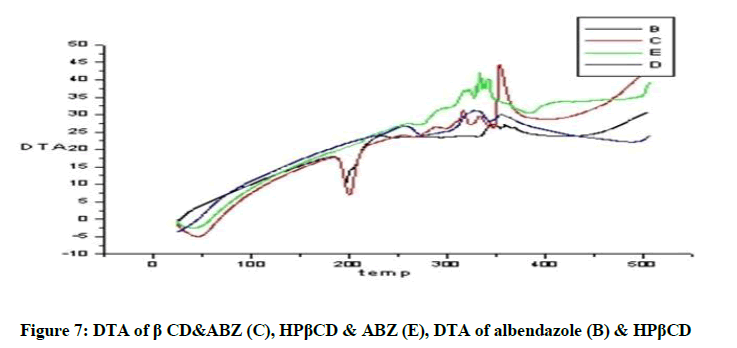
Figure 7: DTA of β CD&ABZ (C), HPβCD & ABZ (E), DTA of albendazole (B) & HPβCD
Comparison of solubility
Solubility comparison of ABZ, ABZ/HPβCD and ABZ/βCD (Kneaded mixture) were done in water. It is found that drug Albendazole is completely insoluble in water and after complexing drug with HPβCD and βCD the wetting property of drug is found to be improved. And from the data obtained ABZ/HPβCD is found to more soluble than ABZ/βCD. Comparison of solubility is shown in Figure 8 and the results are given in Table 5.
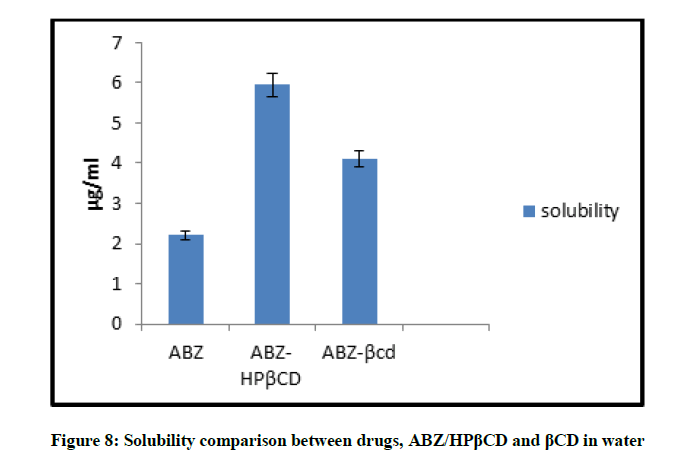
Figure 8: Solubility comparison between drugs, ABZ/HPβCD and βCD in water
| Solubility in Water (µg/ml) | |
|---|---|
| ABZ | 2.208 ± 0.004 |
| ABZ/HBβCD | 5.958 ± 0.001 |
| ABZ/βCD | 4.1 ± 0.003 |
value is expressed as mean ± standard deviation, n=3
Table 5: Comparative solubility data for drug, ABZ/HPβCD and βCD in water
Zeta potential
Zeta potential is commonly used to assess the stability of colloidal systems. Particles with zeta potential more positive than +30 mV or more negative than -30 Mv are normally considered stable. Among the evaluated suspension (ABZ/HBβCD) exhibited maximum zeta potential (Table 6) and is considered to be more stable than other formulations.
| Zeta potential of ABZ-HPβCD and βCD/ABZ Suspension | |
|---|---|
| Suspensions | Zeta Potential |
| ABZ-HPβCD | -52.6 |
| ABZ-βCD | -45.6 |
| Albendazole Suspension | -38 |
Table 6: Zeta potential of ABZ formulations
In vitro drug release studies
The release profile of a drug predicts how a delivery system might function and gives valuable insight into its in vivo behaviour. The release time profile data of Albendazole from HPβCD/ABZ and βCD/ABZ suspensions are represented in the Figures 9 and 10. The release profiles were constructed by plotting the percentage drug release versus time. The highest percent of drug release was observed for formulation ABZ/HPβCD suspension, more than 80% in 90 min, and about 74% in 90 min in case of ABZ/βCD.
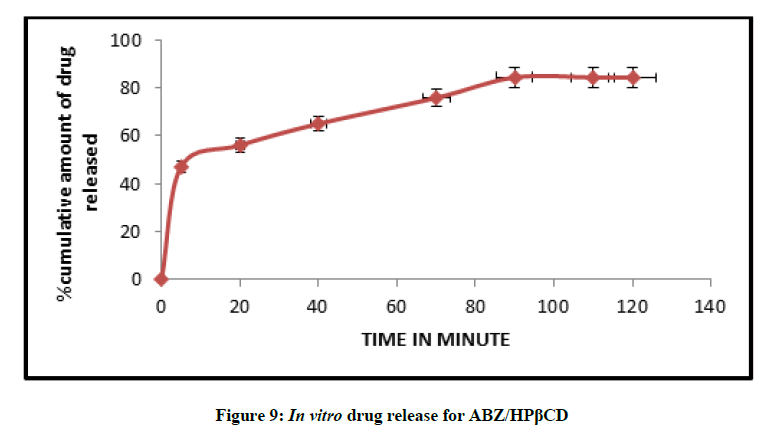
Figure 9: In vitro drug release for ABZ/HPβCD
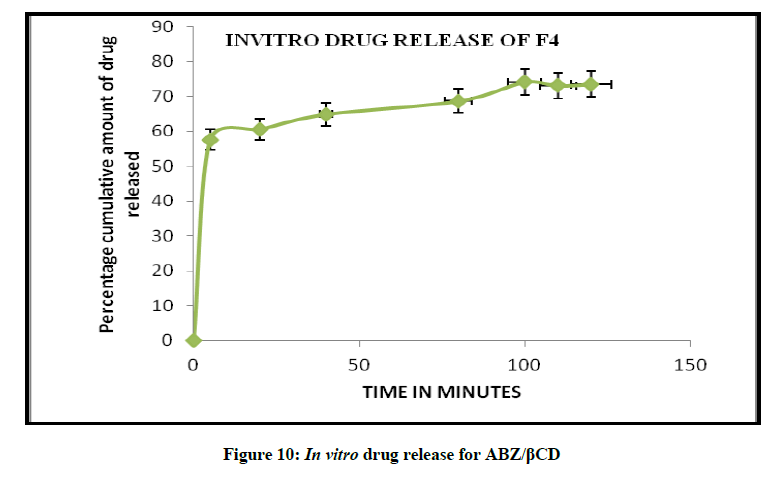
Figure 10: In vitro drug release for ABZ/βCD
Mathematical modelling
The curvilinear nature of the cumulative percentage drug release versus time plots suggest that the drug release from the complexes does not follow zero order kinetics. The data modeling are summarized in Figures 11 and 12 and this observation is supported by low value of correlation co efficient obtained in all case where the dissolution data were fitted to zero order model. The in vitro diffusion studies confirmed that drug release was governed by Higuchi Kinetics, and the release kinetics of drug was found to be first order. Higuchi’s diffusion model gave a better and best fit of release data indicating diffusion dominating as the plots showed maximum linearity with R2 value of 0.980 for ABZ/HPβCD than ABZ/βCD. The korsmeyer Peppas model was found to be linear with the correlation coefficient value of 0.967 and n value for ABZ/HPβCD was 0.197. The n values from Koresmeyer Peppas model of most of the formulation is less than 0.5 indicating that the release pattern follows mainly fickian diffusion mechanism.
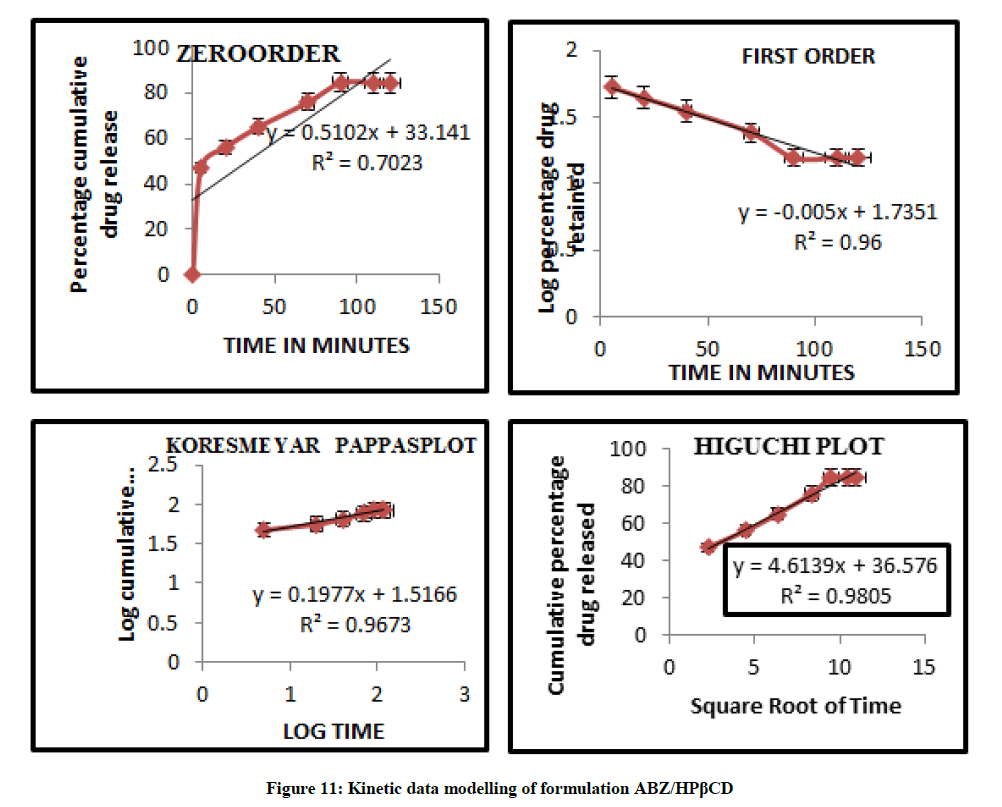
Figure 11: Kinetic data modelling of formulation ABZ/HPβCD
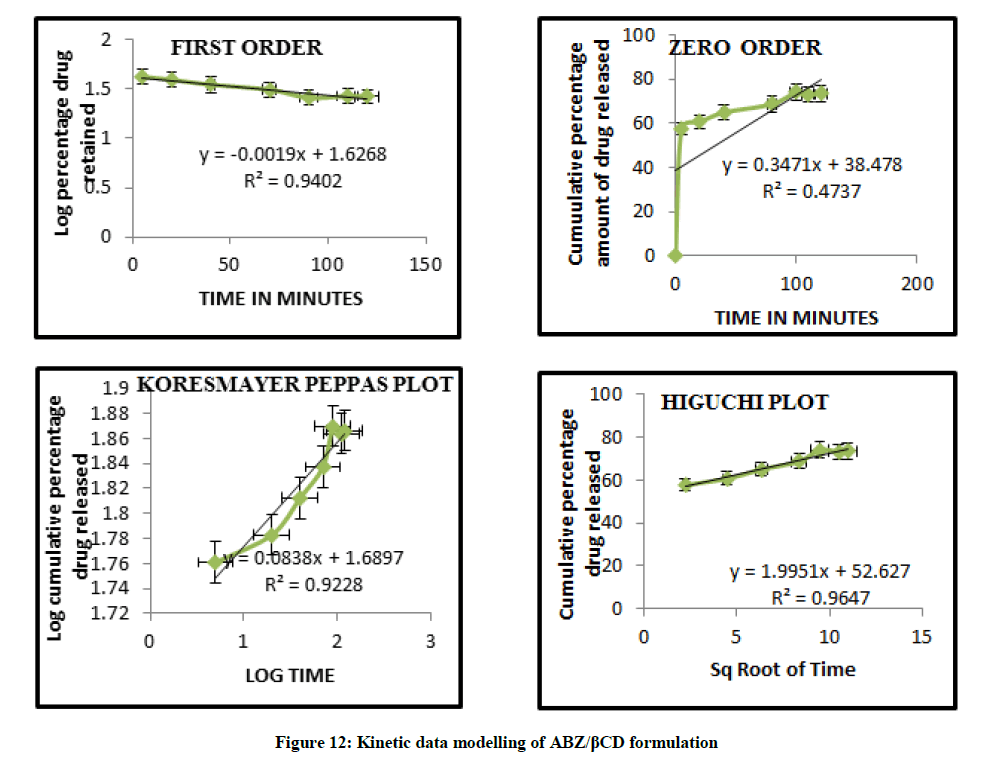
Figure 12: Kinetic data modelling of ABZ/βCD formulation
Therefore ABZ/HPβCD is said to be optimized formulation because,
❖ It gave good zeta potential value.
❖ It showed more than 80% of drug release in 90 min
❖ The wetting property of drug is found to be increased after complexing with HPβCD.
Kinetic Modelling of ABZ/HPβCD
Liver enzyme analysis
The present studies were undertaken to evaluate the metabolic changes that could be encountered in response to Albendazole administration. The results are given in Tables 7 and 8. From the results is found that the activities of AST and ALT is increased in the case of ABZ/βCD formulation but in the case of ABZ/HPβCD formulation there is no increase in the activity of AST and ALT when compared to control group. The activity of ALP is found to be increased in both ABZ/βCD and ABZ/HPβCD when compared to the control. The present results were also in agreement with Davis et al (1989) who stated that most common side effect of Albendazole is the increase in the level of AST, ALT and ALP and drug is not recommended for patients with hepatic cirrhosis [16,17].
| Formulation | Correlation Coefficients | Korsmaeyr Equation (n values) | |||
|---|---|---|---|---|---|
| Zero order Equation | First order Equation | Higuchi Equatin | Korsmaeyr Equation | ||
| ABZ/βCD | 0.473 | 0.94 | 0.964 | 0.922 | 0.083 |
| ABZ/HPβCD | 0.7023 | 0.96 | 0.9805 | 0.9673 | 0.197 |
Table 7: Kinetic data modelling of different formulations of ABZ
| Hepatic function panel | Control | Marketed | ABZ/HPβCD | ABZ/βCD |
|---|---|---|---|---|
| AST(SGOT)-Serum | 100.7 IU/L | 133.6 IU/L | 99.0 IU/L | 110.1 IU/L |
| ALT(SGPT)-Serum | 16.7 IU/L | 18.6 IU/L | 5.0 IU/L | 12.5 IU/L |
| ALP(Alkalinephosphatase) | 294.9 IU/L | 703.8 IU/L | 511.5 IU/L | 640.6 IU/L |
Table 8: Liver Enzyme Analysis
Conclusion
Present study was to evaluate the metabolic changes that could be encountered in response to albendazole administration. One of the side effects of albendazole, on frequent administration can lead increase in liver enzymes so objective of the study was to find out the effect of albendazole and kneaded mixtures on liver enzymes. The present study reveals that the addition of HPβCD andβCD containing ABZ markedly improves the dissolution properties. By solubility studies it was found that solubility of drug is improved by the addition of cyclodextrins. In the present study binary mixture is formulated into suspension form and optimization was done by in vitro dissolution study and zeta potential measurement helped to find out the stability of both formulations. By conducting in vivo study liver enzyme analysis also performed to find out any elevation in liver enzymes like alanine amino transferase, aspartateamino transferase and alkaline phosphatise. From the findings it is concluded that HPβCD/ABZ suspension is having less toxicity compare to the marketed and ABZ-βCD formulations.
References
- S. Torrado, M.L. Lopez, G. Torrado, F. Bolas, S. Torrado, R. Cadorniga, Int. J. Pharmaceut., 1997, 156, 181-187.
- J.A. Castillo, P. Canales, J.J. Garcia, J.L. Lastres, F. Bolas, J.J. Torrado, Drug Develop. Indus. Pharmacy., 1999, 25(12), 1241-1248.
- R.O. Arise, S.O. Malomo, Afr. J. Biochem. Res., 2009, 3(5), 190-197.
- R. Patel, M. Patel, D. Shah, Int. J. Pharm. Wor. Res., 2010, 1(2), 1-21.
- A.C. Tella, Ojeyemi M. Olabemiwo, Musa O. Salawu, Gabriel K. Obiyenwa, Int. J. Physical Sci., 2010, 5(4).
- N.C.T. Cavalcanti, G.D. Sousa, M.A.M. Tabosa, J.L.S. Sobrinho, L.B. Leal, D.P. de Santana, Brazilian J. Pharmaceut. Sci., 2012, 48(2).
- S. Tripathy, P.K. Kar, A.A. Majeed, Int. J. Pharma Bio Sci., 2013, 4(1), 306p.
- N.A. Kasim, M. Whitehouse, C. Ramachandran, M. Bermejo, H. Lennerna, A.S. Hussain, H.E. Junginger, S.A. Stavchansky, K.K. Midha, V.P. Shah, G.L. Amidon, Molec. pharmaceut., 2003, 1, 85-96.
- A.K. Meena, K. Sharma, M. Kandaswamy, S. Rajagopal, R. Mullangi, Acta Pharm., 2012, 62, 563-580.
- V.P. Patel, R.K. Parikh, M.C. Gohel, T.R. Desai, D.R. Bhimani, P.R. Tirgar, Int. J. Pharmaceut. Sci., 2011, 2(1), 161-173.
- F.K. Alanazi, M. El-Badry, M.O. Ahmed, A. Ibrahim, Sci. Pharm., 2007, 75, 63-79.
- S. Dash, P.N. Murthy, L. Nath, P. Chowdhury, Acta Poloniae. Pharmaceutica. Drug. Res., 2010, 67(3), 217-223.
- S. Tripathy, P.K. Kar, A.A. Majeed., Int. J. Pharma Bio Sci., 2013, 4(1), 306-319.
- C. Trandafirescu, A. Gyeresi, M. Kata, Z. Aigner, Z. Szabadai, Timisoara Med. J., 2007, 57(1).
- M.A. Abd El-Rahman, I.M. Adbel-Nabi, M.A. Omran, M.F. Mohamed, Egyptian J. Biol., 1999, 1, 19-29.
- M. El-Badry, Bull. Pharm. Sci., 2008, 31, 123-135.
- P. Madhulita, P. Ganesh, M. Amol, M. Sunil, M.E. Rao Bhanoji, Int. J. Drug Dev. Res., 2010, 2(4), 669-675.



