Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 8
Identification of Impurities and Improved the Synthesis of Lacosamide
Srinivasachary K*, Subbareddy D, Shankar BR, Narayan GKSS, Shashikant T, Somannavar YS and Ramadevi B
Chemical Research and Development, Aurobindo Pharma Ltd, Survey No.71 and 72, Indrakaran (V), Sangareddy District 502329, Telangana, India
- *Corresponding Author:
- Srinivasachary K
Chemical Research and Development
Aurobindo Pharma Ltd, Survey No.71 and 72, Indrakaran (V)
Sangareddy District 502329, Telangana, India
Abstract
The three step process used in the synthesis of Lacosamide (1) was studied and optimized leading to a more robust process. The key process improvements identified and included implementation of telescopic process improvements identified and incuded extraction and solvent exchange and significant optimization of final reaction including the beneficial effect of using aqueous ammonia resulting zero ppm level of residual reagent of dimethyl sulfate.
Keywords
Boc-D-Serine, Benzyl amine, Acetic anhydride, Dimethyl sulfate.
Introduction
Lacosamide is designated chemically as (R)-N-Benzyl-2-acetamido-3-methoxy-propianamide (1) is an antiepileptic drug. It is also useful in the adjunctive treatment of partial-onset seizures and neuropathic pain. It is functionalized amino acids that produce activity in maximal electroshock seizures test. It is believed to act through voltage-gated sodium-channels without effecting the fast inactivation of voltage gated sodium channels resulting in stabilizationzation of hyperexitable neuronal membranes and inhibition or repetitive neuronal firing. Lacosamide (Figure 1) is marketed under the trade name Vimpat® [1].
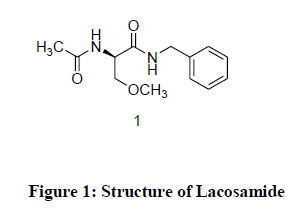
Figure 1: Structure of Lacosamide
Materials and Methods
Experimental
1H-NMR spectra recorded on Bruker 300, variant 500 spectrometer at 300 and 500 MHZ and chemical shifts were reported in parts per million relative internal standard. Infrared (IR) Spectra were recorded in the solid stage as KBr dispersions using perkin-elmer spectrometer. Mass spectra were recorded on API2000 perkin-elmer PESCIEX mass spectrometer. The melting points were recorded on open capillaries and are uncorrected [2]. The experimental procedure is described as below (on the basis of Scheme 4) and according to Scheme 4; Boc-D-Serine (2) is the key raw material for the preparation of Compound (1).
Synthesis of (R)-N-Benzyl -2-amino-3-Hydroxy propanamide (13)
Boc-D-Serine(2) (100 g, 0.4873 mole) was suspended in Methylene chloride (600 ml), cool to below 5°C. N-methyl morpholine (51.75 g, 0.511 mole) was added in 5-10 min at below 5°C, resulting mixture was stirred to get clear solution. The solution was added to a mixture of Isobutyl chloroformate (69.88 g, 0.511 mole) in Methylene chloride (600 ml) at -15°C and agitate the reaction mass for 1 h results the formation of mixed anhydride unstable intermediate [3].
Benzyl amine (54.82 g, 0.511 mol) was added at 5°C-10°C and agitate the reaction mass for 1 h and successively wash the reaction mass with D.M.Water, Aqueous acetic acid to produce (R)-N-Benzyl -2-N-Boc-amino-3-Hydroxy propanamide (11) in Methylene chloride and contain 93.31% purity by qualitative HPLC analysis. Concentrate the organic layer to reach reaction mass volume 3 times [4-7].
Con. HCl (35% w/w, 112.5 Ml, 1.22 mole) was added to partially conc. mass (11) at 25°C-30°C and maintained 1 h and wash the reaction mass DM water, followed by solid sodium-chloride (60 g, 1.025 mole) at 25°C-30°C and adjust the pH to 11 with 50% w/w sodiumhydroxide solution results precipitated product. Filtered and washed with chilled Methylene chloride to give a white solid (66 g, 70%) HPLC purity: 99%, M. P.: 95-97°C; IR (KBr, Vmax, cm-1): 3294, 2923, 2876, 1631; 1H-NMR (300 MHz, DMSO-d6, δ ppm): 1.85 (brs, 2H), 3.25-3.28 (t, 1H), 3.40 (d, 2H), 4.23-4.25 (S, 2H), 4.29-4.85 (brs, 1H), 7.27.3 (m, 5H), 8.39 (brs, 1H); Mass ESI: m/z: 195 [M+H]+, C10H14N2O2, Calculated 194.
Synthesis of (R)-N-Benzyl -2-Acetamido-3-Hydroxy propanamide (5)
Compound (13) (100 g, 0.515 mol) was suspended in Methylene chloride (1000 ml), cool to below 10°C, Acetic anhydride (57.8 g, 0.566 mol) was added and agitate for 2 h at below 10°C, Toluene was added at below 10°C, Filtered and washed with Toluene to give white solid (97.3 g, 80%), M. P.: 146-148°C; IR (KBr, Vmax,cm-1): 3294, 3087, 3027, 1642, 1631, 3351; 1H-NMR (300 MHz, DMSO-d6, δ ppm): 1.87 (s, 3H), 3.58 (t, 2H), 4.28 (m, 3H), 4.93 (t, 1H), 7.21-7.30 (m, 5H), 7.96 (d, 1H), 8.38 (t, 1H); Mass ESI: m/z: 237 [M+H]+, C12H16N2O3, Calculated 236.
Synthesis of (R)-N-Benzyl -2-Acetamido-3-Methoxy propanamide (1)
Compound (14) (100 g, 0.423 mole) was suspended in aqueous 1,2-Dimethoxy ethane (1500 Ml) at 25°C-30°C, cool to 0°C, Dimethyl sulfate (117.5 g, 0.931 mol added at 0°C, aqueous 20%w/w sodium hydroxide solution (sodium hydroxide 28.66 g, 0.719 mole) in DM water (143 g) was added. Stirred the reaction for 12 h separated the layers, organic layer was washed with aqueous ammonia followed by saturated brine solution. Concentrated the organic layer at 30°C-40°C and dissolved the conc. mass in Methylene chloride (1000 ml) and washed with DM water, concentrate the organic layer results solid material was obtained.
Suspend the solidmass in Isopropyl acetate (400 ml) and heat to 50°C for 1h and allowed to cool to 25°C filtered and washed with Isopropyl acetate to give white solid (70 g, 66%); M. P. :143-145°C; IR (KBr, Vmax,cm-1): 3294, 3087, 3027, 2923, 2876; 1H-NMR (300 MHz, DMSO-d6, δ ppm): 1.96 (S, 3H), 3.34 (s, 3H) 3.46 and 3.74 (m, 2H), 4.43 (m, 2H), 4.62 (m, 1H), 6.79 (d, 1H), 7.22-7.48 (m, 5H); Mass ESI: m/z: 237 [M+H]+, C13H18N2O3, Calculated 250.
Boc-D-Serine (2)
IR (KBr, Vmax, cm-1): 3294, 1642, 1631, 3000, 3600, 1398, 1372; 1H-NMR (300 MHz, DMSO-d6 δ ppm): 1.39 (S, 9H), 3.62 and 3.64 (d, 2H), 3.4 (t, 1H), 6.71 (b, 1H), 11.69 and 12.79 (b, 1H); Mass ESI: m/z: 204.1 [M-H]-, C8H15NO5, Calculated 205.
(R)-N-Benzyl-Boc-3-Hydroxy Propanamide (11)
IR (KBr, Vmax, cm-1): 3294, 1642, 1631, 1120, 3087, 3027, 1389, 1366; 1H-NMR (300 MHz, CDCl3 δ ppm): 1.463-1.567 (S, 9H), 3, 1 (d, 1H), 4.3 (m, 2H), 4.5 (2d, 2H), 4.14 (t, 1H), 5.6 (d, 1H), 7.04 (t, 1H); Mass ESI: m/z: 295, M+H]+, C15H22N2O4, Calculated 294.
Results and Discussion
Some problems were encountered in the synthesis of Lacosamide (1) in the first generation process (Scheme 1) involving the D-Serine (3) is treated with acetic anhydride, acetic acid to yield (R)-2-acetamido-3-hydroxy propionic acid (4). Compound (4) is undergoes amidation with Benzyl amine to produce (R)-N-Benzyl-2-acetamido-3-hydroxy propanamide (5), further Compound (14) involves in O-methylation with Methyl iodide, Silver oxide to produce Compound (1) [8].
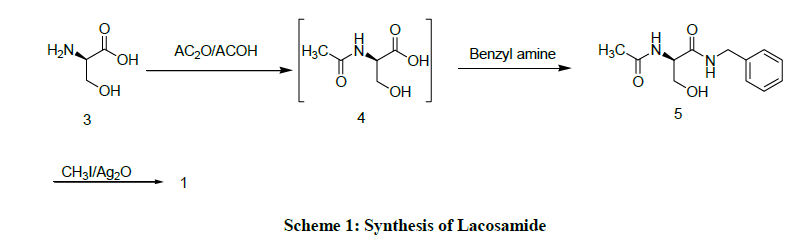
Scheme 1: Synthesis of Lacosamide
The disadvantageous in the preparation of Compound (1) with the above process it contains various impurities which must be removed by column chromatography and employing the column chromatography technique is tedious and laborious and also involves use of large quantities of solvents.
Similarly, the Scheme 2 process for preparation of Compound (1) by reacting D-Serine (3) with Benzyl chloroformate in the presence of Magnesium-oxide to produce Cbz-D-Serine (6) which is further reacted with Benzyl amine in the presence of N-methyl morpholine, Isobutyl-chloroformate in dry Tetrahydrofuran to produce (R)-N-Benzyl-2-(Carbo benzyloxy amino)-3-hydroxy propanamide (7). The Compound (7) is reacted with Methyl iodide in the presence of Silver-oxide to produce (R)-N-Benzyl-2(Carbo benzyoxy amino)-3-methoxy- propanamide (8). The Compound (8) undergoes hydrogenation in the presence of palladium catalyst to produce (R)-N-Benzyl-2-amino)-3-methoxy propanamide (9). Further Acetylation of Compound (9) with acetic anhydride in the presence of Pyridine in Tetrahydrofuran to produce Compound (1) [9].
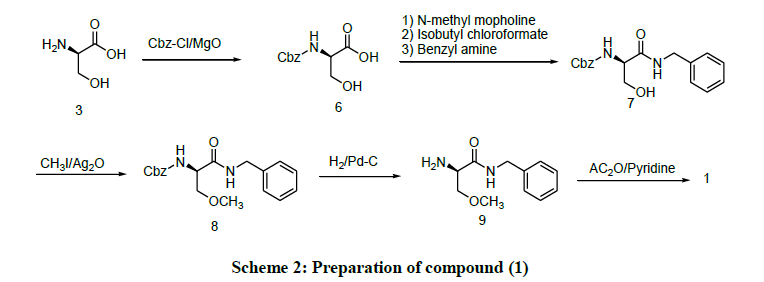
Scheme 2: Preparation of compound (1)
The disadvantage of above process is the use of Silver-oxide in the O-methylation step this reagent is highly expensive and also results in partial racemisation which reduce the yield.
To overcome the above problems the present work describes two schemes (Schemes 3 and 4) to improve the process for preparation of Compound (1) based on our patents 1,2,3.
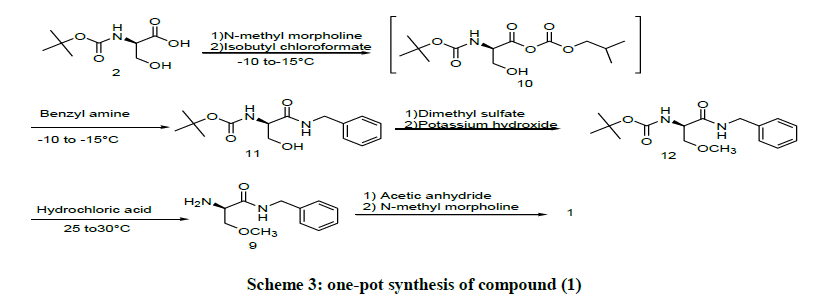
Scheme 3: one-pot synthesis of compound (1)
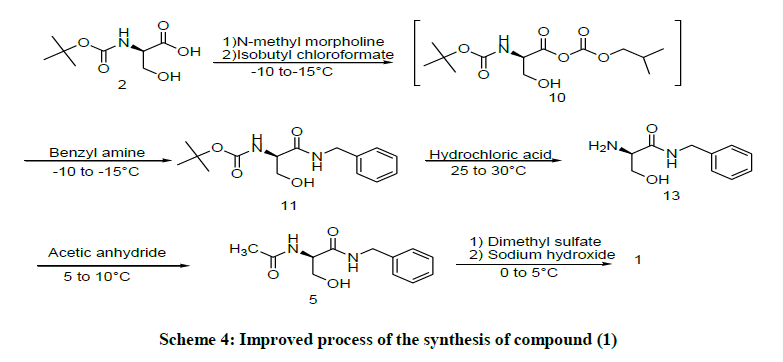
Scheme 4: Improved process of the synthesis of compound (1)
Scheme 3 provides a one–pot process for preparation of compound (1), and it comprises the Boc-D-Serine (2), with Benzyl amine in the presence of a base N-methyl morpholine and an activator of carboxylic group with Isobutyl chloroformate at -10°C to -15°C to produce (R)-N-Benzyl- 2-N-Boc-amino-3-Hydroxy propanamide (11). In this process before adding of Benzyl amine there is a formation of unstable mixed anhydride (10), which is highly moisture sensitive, So for that it is need to apply the the nitrogen atmosphere [10]. The compound (11) in situ undergoes O-methylation with Dimethyl sulfate, potassium hydroxide at 0°C-5°C, to form (R)-N-Benzyl-2-N-Boc-amino-3-methoxy propanamide (12) and reaction completes in 8 h and reaction mass was washed with aqueous ammonia to remove unreacted Dimethyl sulfate, followed by washed with DM water, evoparated the organic layer under reduced pressure to reach three volumes and compound (12) at in situ to react with 35%w/w Hydrochloric acid and agitate for 2 h results in the formation of (R)-N-Benzyl-2-N-amino-3-methoxy propanamide (9). Compound (9) is at in situ react with acetic anhydride by adding a catalytic amount of N-methyl morpholine at 0°C-5°C and then allowed to 25°C-30°C, stirred the reaction mass for 1 h to complete the reaction. After completion of the reaction reaction mass to be worked up and isolated the compound (1) by crystallizing from Isopropyl acetate with 49% yield [11].
In continuous further developed the another improved process Scheme 4 process for to increase the more yield of compound (1) with high purity. The Boc-D-Serine (2) is converted into (R)-N-Benzyl-2N-Boc-amino-3-Hydroxy propanamide (13), in two reaction sequence involving amidation followed by deprotection of Boc group.
In this process compound (2) is treated with Isobutyl chloroformate and N-methyl morpholine in dichloromehane to form mixed anhydride (10) and it is immediately reacts with Benzyl amine to form (R)-N-Benzyl-2-N-Boc-amino-3-Hydroxy propanamide (11). The compound (11) at in situ is under went Boc Deprotection to form (R)-N-Benzyl-2-N -amino-3-Hydroxy propanamide (13), it was isolated in solid state [12].
The compound (13) under goes acetylation with acetic anhydride with using of Methylene chloride, Toluene results the isolated white solid material i.e. (R)-N-Benzyl-2-acetamido-3-hydroxy propanamide (5). The compound (5) is under goes O-methylation with using of Dimethylsulfate, sodium hydroxide leads to form compound (1) with high purity and high yield.
It is observed that Scheme 4 is industrially viable process in comparison with Scheme 3 and gives higher yield. During the development of Scheme 4 we have studied different reaction conditions as described below.
*Reaction conditions: 2 mole equivalent reagents, 1.7 mole equivalent Base, Solvent: Methylene chloride (10 vol) at 0°C-5°C Table 1 shows that Dimethyl sulfate,sodium hydroxide and solvent Methylene chloride at 0°C-5°C condition provides higher conversion to compound (1) in comparison with other reagents,bases with Methylene chloride at 0°C-5°C. And apart from Methylene chloride, we have screened various solvents like Acetone, Acetonitrile, 1,2-Dimethoxy ethane to convert compound (5) to compound (1), among in those solvents 1,2-Dimethoxy ethane provided a significant higher conversion (Table 2).
| S. NO. | Reagents and Bases | Conversion [by HPLC (area%)] | |
|---|---|---|---|
| Compound (1) | Compound (5) | ||
| 1 | Trimethyl sulfoxonium iodide/Sodium hydroxide | 0.67 | 78.8 |
| 2 | Dimethyl sulfate/Sodium hydroxide | 90 | 9 |
| 3 | Methyl Iodide/Sodium hydroxide | 46 | 33 |
| 4 | Dimethyl sulfate/Potassium hydroxide | 77 | 3.9 |
Table 1: Conversion of Compound (5) to Compound (1)
| Solvent | Compound (1) | Compound (5) |
|---|---|---|
| Acetone | 85% | 4.45% |
| 1,2-Dimethoxy-ethane | 93% | 0.67% |
| Methylene chloride | 90% | 1.5% |
Table 2: Conversion of compound (5) to compound (1) in different solvents
During the synthesis of Compound (1), if it contains traces of Dimethylsulfate it leads to advers effects. To avoid the traces of Dimethyl sulfate we are using dilute aqueous ammonia, followed by DM water wash results in the isolated compound (1) contains zero ppm level of Dimethyl sulfate. If the isolated Compound 1 is not pure it may contains impurities such as (R)-N-Benzyl-2-amino)-3-methoxy propanamide 9, (R)-N-Benzyl- 2-acetamido-3-hydroxy propanamid 5, (R)-N-Benzyl-2-acetamido-3-acetoxy-propanamide (14), and these impurities conformed by NMR, Mass analysis. The impurity (9) is originated in final stage it is due to degradation of Compound (1), To control such impurity we have to maintain the reaction of O-methylation in the final stage at 0°C-5°C.
The impurity (5) is appearing in Compound (1) is due to incompletion of O-methylation in final stage, to avoid such impurity it is require to sufficient reagent Dimethyl sulfate and Base sodium hydroxide for to completion reaction.
The impurity (14) is originated in the acetylation of (R)-N-Benzyl-2N-Boc-amino-3-Hydroxy propanamide (13), where if excess reagent of Acetic anhydride it participates in acetylation leads to form impurity (14). To avoid formation of such impurity, we have to use the reagent Acetic anhydride it must be in 1: 1.05 mole equivalent with respect to compound (13).
(S)-N-Benzyl-2-acetamido-3-methoxy propanamide 16, it is enantiomer of Compound (1), the formation of enantiomer is very rare if unless and until the Compound (2) contains traces of Boc-L-Serine then it is carry over in the formation of Compound (1). To avoid such enantiomer we must use the 100% chiral purity of compound (2).
The impurity (15) (Scheme 5) i.e (R)-N-Benzyl-2-acetamido-3(methoxy-methoxy)-propanamide was identified based on LC-MS studies and synthesized with using (R)-N-Benzyl-2-acetamido-3-hydroxy propanamide (5), Methane sulfonic acid, Dimethoxy methane, 1,2-Dimethoxy ethane and isolated the product with using of prep. HPLC and conformed by 1H-NMR, Mass analysis.

Scheme 5: Synthesis of impurity (15)
We found the route cause of impurity (15), in Isolated Compound (1), it is due to from mild acidic nature of 1,2-Dimethoxy ethane. To overcome the formation of impurity, we use aqueous 1,2-Dimethoxy ethane, resulting the formation of Compound 1 it contains not detected level of Impurity (15).
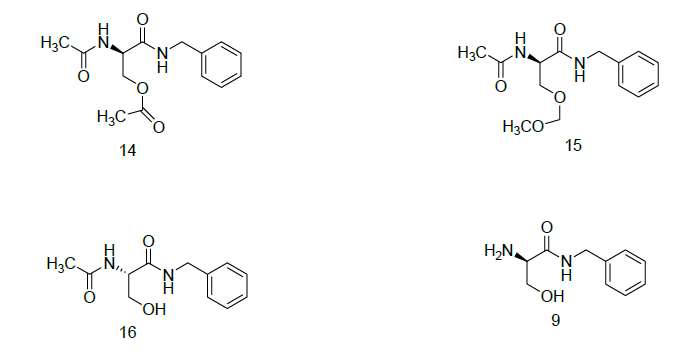
(R)-N-Benzyl 2-acetamido-3-acetoxy propanamide (14)
IR (KBr, Vmax,cm-1): 3294, 3087, 3027, 2923, 2876, 1642, 1631, 1750-1735, 1776, 1614, 1522, 1497, 1455, 1138; 1H-NMR (300 MHz, DMSO-d6, δ ppm): 1.88 (s, 3H), 1.98 (s, 3H), 4.16 (d, 2H), 4.29 (m, 2H), 4.57 (q, 1H), 7.2-7.34 (m, 5H, ArH), 8.24 (d, 1H), 8.61 (t, 1H); Mass ESI : m/z: 279 [M+H]+, C14H18N2O4, Calculated: 278.
(R)-N-Benzyl-2-acetamido-3(methoxy-methoxy)-propanamide (15)
IR (KBr, Vmax,cm-1): 3294, 3087, 3027, 2923, 2876, 1642, 1631, 1750-1735, 1120; 1H-NMR (300 MHz, CDCl3, δ ppm): 2.03 (S, 3H), 3.6 and 3.96 (2m, 2H), 4.47 (d, 2H), 4.57-4.65 (m, 3H), 6.48 and 6.67 (2brs, 2H), 7.27-7.35 (m, 5H); Mass ESI: m/z: 281.2 [M+H]+, C14H20N2O4, Calculated: 280.
(R)-N-Benzyl-2-amino-3-methoxy propanamide (9)
IR (KBr, Vmax, cm-1): 3351, 3294, 3087, 3027, 2923, 2876, 1120, 1614, 1522, 1497, 1455, 1138; 1H-NMR (300 MHz, CDCl3, δ ppm): 3.25 (s, 3H), 3.38-3.45 (m, 3H), 4.28-4.31 (m, 2H), 7.21-7.33 (m, 5H), 8.41 (t, 1H); Mass ESI : m/z :209 [M+H]+, C11H16N2O2, Calculated: 208.
(S)-N-Benzyl-2-acetamido-3-methoxy propanamide (16)
IR (KBr, Vmax, cm-1): 3294, 3087, 3027, 2923, 2876, 1642, 1631, 1614, 1522, 1497, 1455, 1138; 1H-NMR (300 MHz, DMSO-d6, δ ppm): 1.86 (s, 3H), 3.25 (s, 3H) 3.46 & 3.74 (2 m, 2H), 4.27 (d, 2H), 4.43 (m, 2H), 4.47 (m, 1H), 7.2-7.32 (m, 5H), 8.08 (d, 1H), 8.48 (t, 1H); Mass ESI: m/z: 251.3 [M+H]+, C13H18N2O3, Calculated: 250.
Conclusion
To summarize we have improved the synthesis of Lacosamide (1) with high purity and good yield from easily available starting materials.
Acknowledgement
The authors thank to Aurobindo pharma Ltd. for supporting this work. The authors are also thankful to collegues at the analytical research development and chemical research department of Aurobindo Research Centre for the co-operation and fruitful discussions.
References
- K.A.S.S. Narayan, G.D. Subba Reddy; K. Srinivasachary, I. Aminul, Sivakumaran, M. Sundaram, From Indian Pat. Appl, 2014, IN 2010CH01382 A2.
- K.A.S.S Narayan, G.D. Subba Reddy, B.S. Reddy, K. Srinivasachary, K. Gowrisankar Rao, Yatcherla Srinivasa Rao, I. Aminul, Sivakumaran, Meenakshi Sundaram, From PCT int Appl, 2011,WO 2011/144983A2.
- K.A.S.S. Narayan, G.D. Subba Reddy, B.S. Reddy, K. Srinivasachary, K. Gowrisankar Rao, Yatcherla Srinivasa Rao, I. Aminul, Sivakumaran, Meenakshi Sundaram, From PCT US 2013/0123537A1.
- H. Zhenyou, Y. Shangyan, W. Limin, C. Defeng, L. Liangzhe, X. Qingzheng, S. Li, From Faming Zhuanti Shenqing, 2015, CN 104892450 A2.
- M. Alain, B. Didier, V. David, C. Nicolas, From PCT, WO212/041986A1.
- R. mangesh, N. Nair, R. Shrigadi, N. Balkrishna, P. Aditi Milind, From PCT, WO212/014226A1.
- M.M. Kumar Singh, P. Kumar, K. Chandra Has, From US. Pat. Appl. Publ., 2009, US 20090143472A1.
- B. Pandey, K. Shah, From PCT int. Appl., 2011, WO 2011039781A1.
- M.P. Reddy, V. Madalapu, N.V. Chowdary, From PCT. int. Appl., 2011, WO 20110811A1.
- B. Alberto, C. Patrizila, V. Domenico, B. Giorgio, PCT. int. Appl, 2011, WO 2011092559A1.
- J. Desai, S. Prabhurao, B. Sandipan, Vijay kumar, A. Girish, L. Tarde, D. Nusa, D. Sagharakshit From Indian Pat. Appl, 2012, IN 2011MU00470A2.
- B. Vazquez, P. Miguel, L. Seres, J. Miguel, C. Martinez, Y.M. Antonieta, J. Lagunas, J. Alberto, S. Meerles, Donato, V. Miranda, J. Rolando; Z. Huerta, Amando From PCT. int. Appl., 2013, WO 2013030654A1.



