Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 4
In Silico Modeling of RelEB Type II Toxin-antitoxin System in Acinetobacter baumannii as a Therapeutic Target via Antimicrobial Photodynamic Therapy
Maryam Pourhajibagher1, Nava Hosseini2 and Abbas Bahador3,4*
1Dental Research Center, Dentistry Research Institute, Tehran University of Medical Sciences, Tehran, Iran
2Department of Microbiology, Faculty of Biology, College of Science, University of Tehran, Tehran, Iran
3Dental Implant Research Center, Dentistry Research Institute, Tehran University of Medical Sciences, Tehran, Iran
4Department of Microbiology, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran
- *Corresponding Author:
- Abbas Bahador
Dental Implant Research Center
Dentistry Research Institute
Tehran University of Medical Sciences
Tehran, Iran
Abstract
Acinetobacter baumannii is one of the opportunistic bacteria contribute to burn infections. Type II toxin-antitoxin systems (RelEB) are the particular type of toxin-antitoxin modules which take part in different kinds of cellular actions in A. baumannii. Treating burn wounds with antimicrobial agents such as antibiotics forms one of the most recent crucial issues. Recently, Antimicrobial Photodynamic Therapy (aPDT) is considered as the adjuvant and innovative method for conventional antibiotic therapy. In this study, we evaluated the potential of the hierarchical nature of RelE as an antitoxin protein associated with virulence factor in A. baumannii using a number of bioinformatics tools and computer simulation molecular modeling. The predicated structure of RelE exhibited that it is a cationic protein with positive charge (1.1) in pH 7. Random coil was dominate other secondary structures in RelE which was located outside the cell. Based on the results of this study, RelE as a stable protein has a potential site for aPDT.
Keywords
Acinetobacter baumannii, Antimicrobial photodynamic therapy, Bioinformatics, In silico, Toxin-antitoxin system
Introduction
Burn injuries are one the noticeable reasons of death in developing countries [1]. Patients with burn wounds are at high risk of acquiring infections. Acinetobacter baumannii is one of opportunistic pathogens which is associated with nosocomial infections [2]. Acinetobacter spp., are responsible for variety of infections from skin and soft tissue infections to urinary tract infections which specially take place in immunocompromised patients [3]. This opportunistic pathogen is able to acquire fast antibiotic resistance elements, so recently the emergence of extensive drug resistant strains of this bacteria have occurred [4].
Toxin-antitoxin systems are extensively distributed among bacteria and archaea which are associated with antibiotic tolerant cells (persisters). Persister cells may have involvement in chronic and recurrent diseases. They also are implicated in bacterial stress response management [5]. It has been proposed that toxin-antitoxin modules can develop two different processes in bacteria cell; coping with stress by inducing dormant stage or prompting allosteric sacrifice for the population by Programmed Cell Death (PCD) [6].
RelBE is a type II toxin-antitoxin system which is composed of RelE toxin and RelB antitoxin. In favorable environmental conditions, RelB antitoxin can reverse the toxin function like temporary inhibition of cell processes, growth arrest. By inhibition of translation, RelE plays a regulatory role during unfavorable environmental conditions [7].
Although conventional therapies such as using nanoparticle antimicrobial agents and antibiotics can temporarily reduce the count of A. baumannii, this organism cannot be completely eliminated and causing to emerge the antibiotic resistant strains [8,9].
Due to the increasing level of drug resistance, finding new therapeutic approaches and alternatives or supplementary drugs besides antibiotics remains the core priority of scientists worldwide. Antimicrobial Photodynamic Therapy (aPDT) as one of the therapeutic technique can be an effective adjuvant that can overcome the problems caused by the emergence of resistance [10]. During this process, Photosensitizer (PS) dye and visible light are used [11].
According to the literature review, in silico approach enhances our knowledge of the target site to improve the aPDT efficiency [12]. In present study, we used number of bioinformatics tools and computer simulation molecular modeling to predict the hierarchical nature of RelE, as one of the important target for aPDT.
Materials and Methods
Primary sequence analysis
The amino acid sequences of RelE toxin from A. baumanii strain ATCC 19606 with accession number WP_000897309.1 was retrieved from National Center for Biological Information protein database (http://www.ncbi.nlm.nih.gov/protein), in FASTA format. Selected sequence was subjected to Protein- Basic Local Alignment Search Tool (P-BLAST) against Protein Data Bank (PDB) (http://blast.ncbi.nlm.nih.gov/Blast.cgi) to retrieve and find similar sequence to RelE.
Secondary and tertiary structure prediction
The secondary protein structure was predicted by self-optimized prediction align method, SOPMA (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html) using default parameters (Window width: 17, similarity threshold: 8 and number of states: 4) and European Bioinformatics Institute (EMBL-EBI; https://www.ebi.ac.uk/services). The three dimensional structure of RelE was obtained from https://www.ncbi.nlm.nih.gov/Structure/VAST and interactions of RelE molecule were investigated.
Prediction of physicochemical properties and functional characterization of RelE
The quality of RelE structure was validated by means of programs PROCHECK, ERRAT and VERIFY-3D that are available at the Structure analysis and Verification server (http://nihserver.mbi.ucla.edu/SAVES/).
Accordint to the previous in silico studies, ProSA-web server (https://prosa.services.came.sbg.ac.at/prosa.php) was used to determine the interaction energy per residue and confirmation of the local and overall models quality of RelE [13-17].
Analysis tools for determination of physicochemical properties including Isoelectric Point/Molecular Weight (pI/MW), a tool for predicting RelE pI and MW, ProtScale for amino acid scale representation such as hydrophobicity plots, ProtParam to assess numerous physicochemical parameters, peptide mass as a tool for theoretically cleaving RelE and calculating the masses of its peptide and any known processing of cellular or post-translational modifications, were used from the tools page available at UniProt (http://expasy.org) and Expasy ProtParam server (http://www.expasy.org/cgi-bin/protpraram).
Prediction of protein topology and backbone of RelE
The propensities of individual and the conditional probability amino acids to form particular secondary structures were demonstrated using the Garnier-Osguthorpe-Robson (GOR) IV method (https://npsa-prabi.ibcp.fr). The ligand binding site, enzyme commission (EC) numbers and active sites were found by the ITASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER). The Ramachandran diagram that determines which torsional angles can obtain insight into the structure of peptides was plotted via Rampage (http://mordred.bioc.cam.ac.uk~rapper/rampage.php).
The locations of trans-membrane intracellular and extracellular regions of RelE were determined with a method for prediction transmembrane helices based on a hidden Markov model (TMHMM; www.cbs.dtu.dk/services/TMHMM) that is a membrane protein topology prediction method. In addition, the charge of RelE was computed via the protein calculator v 3.4 (http://protcalc.sourceforge.net).
Protein-protein interaction network analysis
The motifs and signal peptides of RelE were analyzed by Motif databases (http://www.genome.jp/tools/motif/) and Interpro server (http://www.ebi.ac.uk/interpro), respectively. STRING (http://string-db.org) quantitatively assimilated the protein- protein interactions network, ranked their significance or validity as targets and gene neighborhood of RelE (http://string-db.org).
Visualization of proteoforms and ligand geometry
Interactive integration of annotated and predicted sequence features together with experimental proteomic evidence were obtained using the Protter server (http://wlab.ethz.ch/protter/). Ligands have suitable pharmacological properties and no reactive functional group. Ligand geometry was determined at the European Bioinformatics Institute (EMBL-EBI) (http://www.ebi.ac.uk) to identify potential ligands which interact with aPDT.
Results
Protein sequence identity analysis at the NCBI GenBank database showed that RelE has 106 amino acids that its estimated structure weight was 12209.01 dalton (average mass) and 12201.43 (Monoisotopic mass). By BLAST, similarity search against RelE identified in A. baumannii displayed that it is similar to the protein structure with accession number of 5JA8. The total score for protein alignment between RelE and 5JA8 was 82.8, with 83% and 43% query cover and identity, respectively. 5JA8 is composed of 6 molecules including 3 molecules of phosphate ion, 1 molecule of acetate ion, 1 molecule of 1,2-Ethanediol and 1 molecule of 4-(2-Hydroxyethyl)-1-Piperazine Ethane sulfonic Acid. The amino acid compositions of RelE in A. baumannii are: Ala (A) 1.9%, Arg (R) 4.7%, Asn (N) 2.8%, Asp (D) 7.5%, Cys (C) 0.9%, Gln (Q) 8.5%, Glu (E) 6.6%, Gly (G) 8.5%, His (H) 0.9%, Ile (I) 8.5%, Leu (L) 9.4%, Lys (K) 10.4%, Met (M) 0.9%, Phe (F) 4.7%, Pro (P) 1.9%, Ser (S) 4.7%, Thr (T) 4.7%, Trp (W) 0.9%, Tyr (Y) 3.8%, Val (V) 7.5%, Pyl (O) 0.0% and Sec (U) 0.0%.
Initial structure prediction showed that RelE had 16 positively charged residues (Arg + Lys) and 15 negatively charged residues (Asp + Glu) in A. baumannii. RelE was a stable protein and was present outside the cell with a grand average of hydropathicity value (GRAVY) −0.466. The very high aliphatic index was 93.68 and instability index (II) was computed to be 31.31, that provides the estimate of the stability of protein in a test tube. RelE theoretical pI was 7.80 and the extinction coefficient in water was 11460 M−1 cm−1 at 280 nm. As you see in Figure 1. The secondary structure prediction defined each residue into either beta strand, alpha helix or random coil structures.
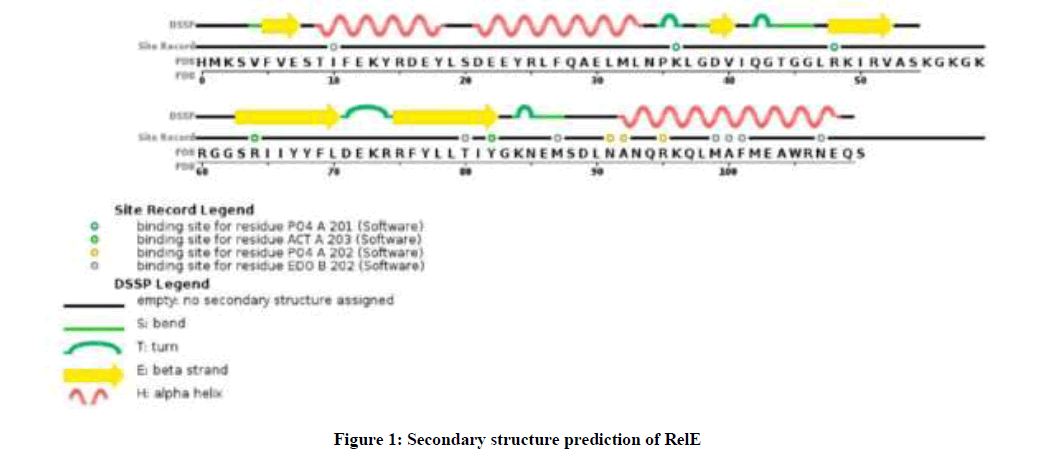
Figure 1: Secondary structure prediction of RelE
GOR IV analysis revealed alpha helix more than random coil or beta strand (Table 1). Three dimensional structures of RelE have been generated by VAST workspace and its quality was confirmed with ProsaWeb server (Figure 2). ProSA output for the RelE shows the local model quality by plotting energies profile of amino acid and bindicating the Z score (or standard score) on a plot based on the entire protein chain in the PDB determined by X-ray crystallography or Nuclear Magnetic Resonance (NMR) spectroscopy (Figure 2b, 2c).
| Groups | Prevalence |
|---|---|
| Alpha helix | 35 is 33.02% |
| 310 helix | 0 is 0.00% |
| Pi helix | 0 is 0.00% |
| Beta bridge | 0 is 0.00% |
| Extended strand | 24 is 22.64% |
| Beta turn | 0 is 0.00% |
| Bend region | 0 is 0.00% |
| Random coil | 47 is 44.34% |
Table 1: The prevalence of different structure in RelE
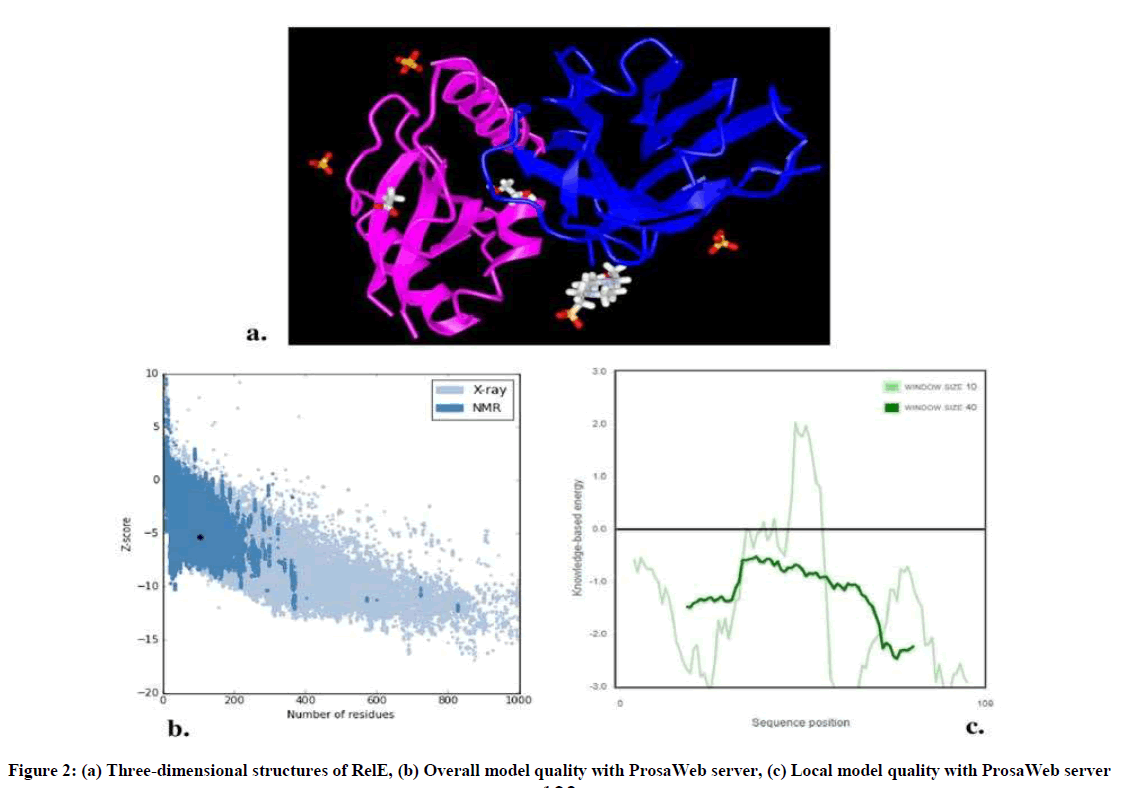
Figure 2: (a) Three-dimensional structures of RelE, (b) Overall model quality with ProsaWeb server, (c) Local model quality with ProsaWeb server
After the analysis performed at the I-TASSER server, it was found that ligand binding site residues were 3, 9, 22, 25, 26, 29, 48, 63, 65, 74 and 76 amino acids with 0.26 C-score. In addition, the active site residue was located at 53 amino acid with 0.496 TM-score, 0.244 C-score, 4.15 RMSD and 0.053 percentage sequence identity in the structurally aligned region. According to the Ramachandran Plot analyses, 98.9% (891/901) of all residues are in favored (98%) regions. As well as, 100% (901/901) of all residues are in allowed (> 99.8%) regions (Figure 3).
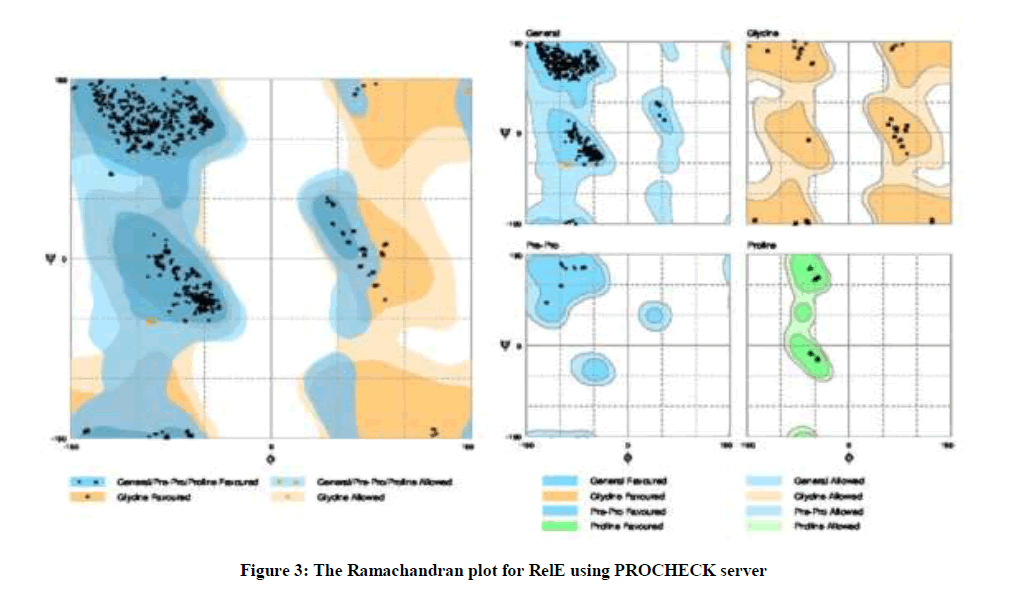
Figure 3: The Ramachandran plot for RelE using PROCHECK server
On the other hand, the results of TMHMM showed that RelE was present outside the cell (Figure 4) and estimated charge of RelE at pH 7.00 was 1.1 by protein calculator.
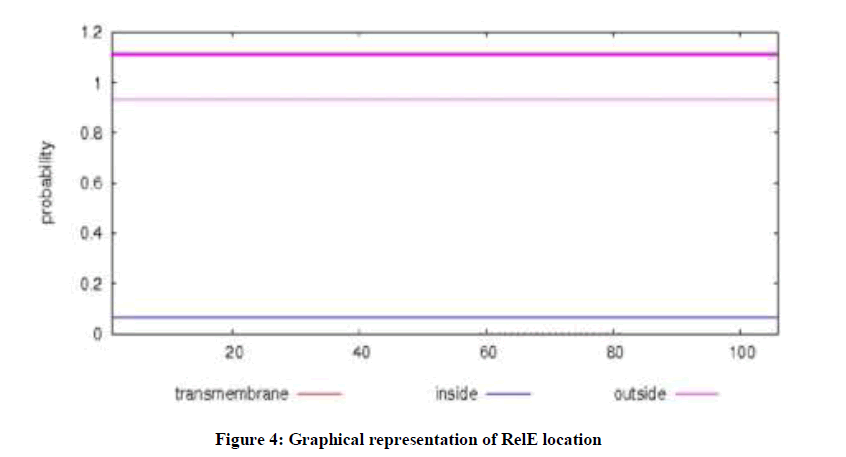
Figure 4: Graphical representation of RelE location
The protein interaction network displays the proteins and their length and type of relationship with RelE. Based on the results, RelE has several predicted functional partners, such as DNA-binding helix-turn-helix protein (F911_03776) with 90 amino acids and score of 0.977, Uncharacterized protein (F911_03778) with 65 amino acids and score of 0.504 and Helicase C-terminal domain protein (F911_01953) with 1025 amino acids and score of 0.424. RelE has motif in position of 41 to 102 amino acids with i-Evalue of 3e-12. Also, there is one N-glyco motif in position of 87 amino acid from RelE (Figure 5).
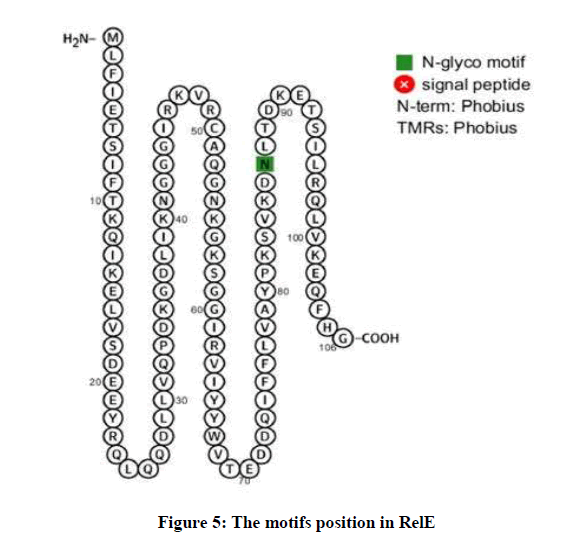
Figure 5: The motifs position in RelE
Discussions
During aPDT, the PS is a nontoxic dye which accumulates and binds to the outer surface and cell wall of microorganisms and then causes the irradiation of the target tissue with light. Therefore, aPDT mediates bacterial cell death by membrane disruption and DNA damage [18]. It has been shown that aPDT not only can be used for killing bacteria, but it can also be used to reduce the impact of bacterial virulence factors and biofilm formation and prevent the colonization of bacteria [19].
A. baumannii as an opportunistic pathogen, with the following characteristics of being Gram-negative, oxidase-negative, non-fermentative, non-motile Coccobacilli has broad range of antibiotic resistance [20-22]. Toxin-antitoxin modules are one of the virulence factor in several microorganisms. As previously mentioned, RelBE is a type II toxin-antitoxin system in A. baumannii which is composed of protein toxin RelE and protein antitoxin RelB [23].
At first, RelBE was discovered on Escherichia coli chromosome, but later it founded on E. coli plasmid either [24]. This module is one of the best studies type II toxin-antitoxin systems. The RelBE encoding locus is common among Eubacteria and Archaea [7]. In A. baumannii, relBE operon encodes RelB-RelE toxin-antitoxin pair which RelB gene was first recognized from mutants with "delayed relaxed" phenotype [25].
This system is actually located RelB and RelE proteins form a non-toxic complex which together with free RelB autogenously regulate the transcription of relBE operon. The relB (antitoxin) gene is located upstream of the relE gene and both are regulated from one mRNA transcript. So the genetic organization of relBE gene pair is characterized like other type II modules [26,27]. RelE toxin in known for cleavage of mRNA positioned at A-site with high codon specificity [8]. Among stop codons cleaved by toxin, UAG is being cleaved with faster rate in comparison to other stop codons like UAA or UGA, while UCG and CAG are cleaved in the most rapid speed among sense codons. By cleaving the mRNAs under inappropriate environmental condition, the level of protein translation is decreased in order to diminish the amino acid consumption under starvation [8]. Under unfavorable conditions, the concentration of RelB antitoxin becomes diminished due to unstableness against Lon protease. By dwindling the RelB concentration, RelE starts to play its toxic role in cytoplasm [28].
As you saw, RelE has 106 amino acids and 6 molecules. Phosphoric acid in RelE is a sequestering agent which binds many divalent cations, including Fe++, Cu++, Ca++ and Mg++ and plays a role in many metabolic processes. As well as, Acetate is a synthetic carboxylic acid with antimicrobial properties. Undissociated acetic acid can enhance lipid solubility allowing increased fatty acid accumulation on the cell membrane or in other cell wall structures and may inhibit carbohydrate metabolism resulting in subsequent death of the microorganism. According to studies, subsequent blocking of RelE toxin transcription and overexpression of RelB antitoxin can restore the protein synthesis and colony formation. So it has been showed that translation blockage by RelE toxin is bacteriostatic rather than bactericidal.
Also it has been showed that RelE toxin can develop rapid adaptation of bacteria to inappropriate growth conditions. In vivo studies has been demonstrated that recovery of protein synthesis after RelE induction is mediated by tmRNA [29]. Taken together, selection target protein structural properties must be visualized to show the effectiveness and the interpretability of the aPDT.
Conclusion
In conclusion, A. baumannii is the chief bacteria involved in life-threatening infection in burn wound patients. In this current study, we demonstrated that RelE can be a suitable target site for aPDT. Nevertheles, more studies are needed to achieve better performance of aPDT against A. baumannii.
References
- M. Pourhajibagher, M. Mokhtaran, D. Esmaeili, A. Bahador, Der Pharmacia Lettre., 2016, 8, 225-229.
- Z. Farshadzadeh, F.B. Hashemi, S. Rahimi, B. Pourakbari, D. Esmaeili, M.A. Haghighi, Front Microbiol., 2015, 6, 1146-1153.
- M.G. Thompson, C.C. Black, R.L. Pavlicek, C.L. Honnold, M.C. Wise, Y.A. Alamneh, Antimicrob. Agents Chemother., 2014, 58, 1332-1342.
- L. Azimi, M. Talebi, M.R. Pourshafie, P. Owlia, A. Rastegar Lari, Int. J. Mol. Cell Med., 2015, 4, 46-53.
- J.A. Lemos, T.A. Brown Jr, J. Abranches, R.A. Burne, FEMS Microbiol. Lett., 2005, 253, 251-257.
- L. Van Melderen, Curr. Opin. Microbiol., 2010, 13, 781-785.
- A. Rheinberg, I.J. Swierzy, T.D. Nguyen, H.P. Horz, G. Conrads, J. Oral Microbiol., 2013, 5, 1-5.
- R. Khajavi, M.M. Shirmohammady Bahadoran, A. Bahador, A. Khosravi, J. Indus. Tex.,2012, 42, 219-230.
- M. Pourhajibagher, N. Chiniforush, R. Raoofian, R. Ghorbanzadeh, S. Shahabi, A. Bahador, Photodiagnosis Photodyn. Ther., 2016, 16, 50-53.
- M. Pourhajibagher, F.B. Hashemi, B. Pourakbari, M. Aziemzadeh, A. Bahador, Open Microbiol. J., 2016, 10, 32-42.
- K. Pedersen, A.V. Zavialov, M.Y. Pavlov, J. Elf, K. Gerdes, M. Ehrenberg, Cell., 2003, 112, 131-140.
- N. Chiniforush, M. Pourhajibagher, S. Shahabi, E. Kosarieh, A. Bahador, J. Lasers Med. Sci., 2016, 7, 76-85.
- M. Pourhajibagher, A. Bahador, Drug Res., 2016, 66, 1-5.
- M. Pourhajibagher, A. Bahador, Photodiagnosis Photodyn. Ther., 2016, 16, 154-160.
- M. Pourhajibagher, A. Bahador, Photodiagnosis Photodyn. Ther., 2017, 17, 98-102.
- M. Pourhajibagher, S. Gharesi, R. Ghorbanzadeh, Der Pharma Chemica., 2015, 7, 243-250.
- M. Pourhajibagher, M.M. Karimi Yazi, A. Bahador, Der Pharma Chemica., 2015, 7, 301-306.
- M. Pourhajibagher, E. Boluki, N. Chiniforush, B. Pourakbari, R. Ghorbanzadeh, M. Azimzadeh, Photodiagnosis Photodyn. Ther., 2016, 15, 202-212.
- M. Pourhajibagher, N. Chiniforush, S. Shahabi, R. Ghorbanzadeh, A. Bahador, Photodiagnosis Photodyn. Ther., 2016, 15, 159-166.
- Z. Oruba, P. Łabuz, W. Macyk, M. Chomyszyn-Gajewska, Photodiagnosis. Photodyn. Ther., 2015, 12, 612-618.
- M. Pourhajibagher, M. Mokhtaran, D. Esmaeili, A. Bahador, Der Pharma Chemica., 2016, 8, 347-351.
- M. Pourhajibagher, H. Kazemian, N. Chiniforush, A. Bahador, Laser Therapy., 2017, 26, 97-103.
- S.K. Christensen, K. Pedersen, F.G. Hansen, K. Gerdes, J. Mol. Biol., 2003, 332, 809-819.
- M. Moritz, P. J. Hergenrother, Proceed. Nat. Acad. Sci., 2007, 104, 311-316.
- V. Tsilibaris, G. Maenhaut-Michel, N. Mine, L. Van Melderen, J. Bacteriol., 2007, 189, 6101-6108.
- G. Hugo, K. Gerdes, J. Mol Biol., 1999, 4, 1401-1415.
- M. Gotfredsen, K. Gerdes, Mol. Microbiol., 1998, 29, 1065-1076.
- P. Pandey, K. Gerdes, Nucleic Acids Res., 2005, 3, 966-976.
- K. Gerdes, K. Susanne, Nature Rev. Microbiol., 2005, 3, 371-382.



