Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 12
Molecular Docking Simulation Study of Novel Flavones and Flavonoids as Aromatase Inhibitors
Sabale Prafulla* and Potey Lata
Department of Pharmaceutical Sciences, Rashtrasant Tukadoji Maharaj Nagpur University, Nagpur-440033, India
- *Corresponding Author:
- Sabale Prafulla
Department of Pharmaceutical Sciences
Rashtrasant Tukadoji Maharaj Nagpur University
Nagpur-440033, India
Abstract
Breast cancer is the most common cancer in women after skin cancer. Breast cancer originates in the breast tissue, most commonly from the inner lining of milk ducts or the lobules that supply the ducts with milk. Breast cancerous cell also depends on estrogen for the growth and development like normal cell, which is called as ER+Ve breast cancer. This study is focus on reduction of level of estrogen and it will be possible by inhibition of estrogen biosynthesis. Aromatase enzyme is essential for the estrogen biosynthesis. In this study, an attempt has been made to design the novel flavones and flavonoids targeting Leu477, Ser478 active binding site of an aromatase using (3EQM) PDB Id for the molecular docking simulations. Schrodinger docking programs was used to compare their suitability for the receptor. Interestingly, in the docking programs, the designed flavonoids have occupied the same binding pocket confirming the selection of active. We have identified a flavonoid which shows docking and Glide score comparatively much higher than those of already reported known inhibitors against aromatase. The protein-ligand complex with top-ranked flavonoid was synthesized by novel designed scheme. The results provide valuable information for structure-based drug design of aromatase inhibitors.
Keywords
Aromatse enzyme, Molecular docking, Imidazole, Breast cancer.
Introduction
Breast cancer is the most common cancer in women after skin cancer. Worldwide, more than one million women developed breast cancer yearly and near about half of these cases has been diagnosed in the Unites States and Europe. Moreover, nearly 40% of the women have been died due to breast cancer. Approximately two-thirds of postmenopausal breast cancer patients have estrogen-dependent breast cancer, which contains estrogen receptors and requires estrogen for tumor growth. Among US women in 2017, there will be an estimated 252710 new cases of invasive breast cancer, 63410 new cases of breast carcinoma in situ, and 40610 breast cancer deaths [1,2]. Heightened awareness of breast cancer risks in the past decades have led to an increase in the number of women undergoing mammography for screening, leading to detection of cancers in earlier stages and a resultant improvement in survival rates. Still, breast cancer is the most common cause of death in women between 45-55 years of age [3].
With advances in screening, diagnosis and treatment, the death rate for breast cancer patients has declined. In fact, about 90% of women newly diagnosed with breast cancer can survive for at least five years. Research is continuously going on to develop even more effective screening and treatment programs [4]. Prolonged exposure to circulating estrogens contributes to increased incidences of breast cancer. Removal of estrogens via oophorectomy decreases the risk of breast carcinoma. In postmenopausal women, large part of estrogens in the breast tissue is derived from in situ biosynthesis using androgens as a substrate. Major sites of estrogen production are ovaries in premenopausal women, placenta in pregnant women and peripheral tissues such as fat, muscles and breast tissues in postmenopausal women [5].
Flavonoids are a group of polyphenolic compounds that occurs naturally in foods of plant origin. It shows a wide variety of biological activities, such as antioxidant, antimicrobial, antiinflammatory and more specific is anticancer activity. It is observed that the Flavonoids moiety possess characteristic pharmacophore pattern (Figures 1 and 2) which is essential for binding to aromatase enzyme and their by its inhibition. Currently non-steroidal aromatase inhibitors possess heterocyclic ring such as imidazole and or triazole act as ligands, substituted aromatic ring at nucleus at suitable position acts as H bond acceptor which bound with Serine-478 present in active site of aromatase enzyme. Basic nucleus plays role as hydrophobic spacer moiety which maintained distance between heme coordinating group and hydrogen bond acceptor moiety [6].
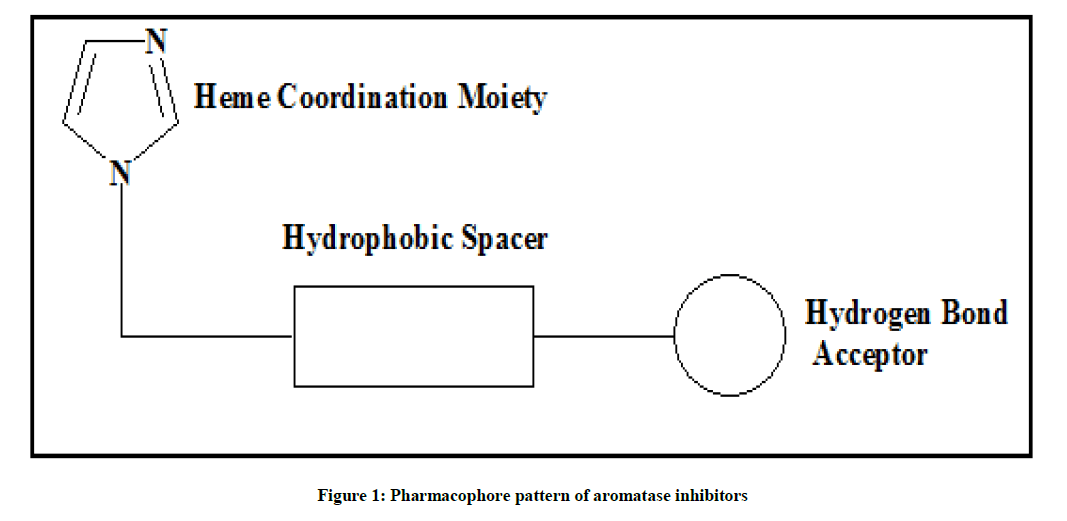
Figure 1: Pharmacophore pattern of aromatase inhibitors
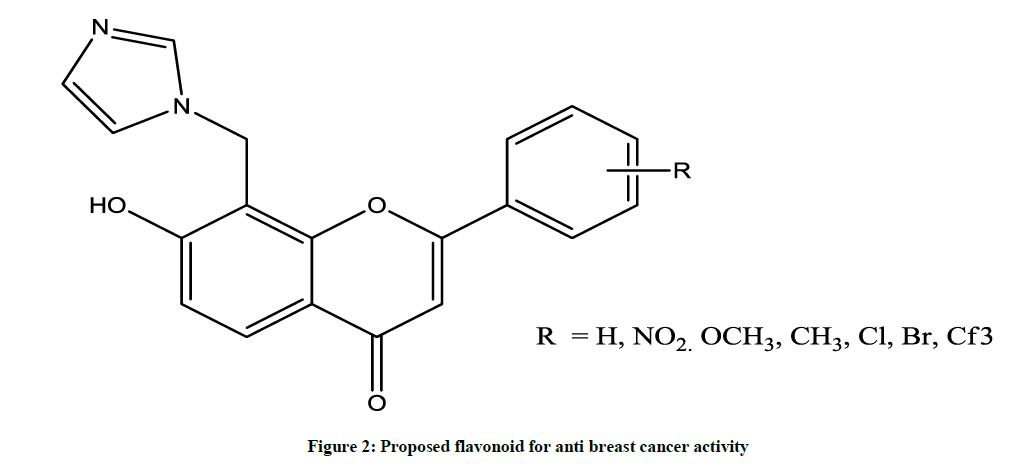
Figure 2: Proposed flavonoid for anti breast cancer activity
This study is based on the computational methods comes under the CAAD. Computer-aided drug discovery (CADD) includes mainly two strategies, which are namely target and ligand based approaches [7]. Target based approach depends on the interaction between structure of target and the ligands on the other hand, ligand-based approach does not need to know the structure of the target and it’s interaction with the ligands but it depends on the structural information and molecular properties of known ligands only. Both these methods have their restrictions [8]. Therefore, to provide better information and to prove their advantages and disadvantages both approaches combine together in order to carry out fundamental screening of a large set of different compounds leading to an unequivocal recognition of a lead compound.
Recently, Ghosh et al. has discovered the crystalline structure of human placental aromatase [6] which is important for the molecular simulation approaches which explain the accessibility of structural facts on the active site of aromatase and helps in understanding the binding interactions of AIs as well as the appraisal of key reaction required in the inhibition of an aromatase. Molecular simulation approaches introduces the molecular basis for the specificity of the aromatase enzyme and its unique catalytic mechanism, which is very important for the development of the next-generation of AIs [9]. Now a days the research is going on to study the chemical reactions and binding mechanisms of a wide range of biological and chemical systems through the Computational chemistry as such, in silico tools such as molecular docking are important for shedding light on the binding modalities and interaction strengths of AIs [10].
Molecular docking simulation study
Ligand preparation
The Schrodinger ligand preparation was done by using LigPrep panel application which consists of series of steps that perform conversion of 2D structures to 3D structure, apply correction to the structure by minimizing the proper bond angles and distances and optimize the structure by minimizing its energy through force-field OPLS3.
Preparation of protein and grid generation
The X-ray crystal structure of an aromatase enzyme with 1.20 Å resolution (PDB ID: 3EQM), [11-13] was obtained from Protein Data Bank (REF). The protein structure was processed by the Protein preparation module of Maestro, v11.5, Schrodinger. In docking study the pre-processing of proteins was carried out by adding hydrogen, creating zero order bonds to metals, creating disulfide bonds, Filling missing side chains using prime, Filling in missing loops using prime, Deleting water beyond 5.00 Ǻ From het group, Generating het state using Epik: PH 7.0+/- 2.
The grid around the receptor was generated using module available with Glide v6.6, Schrodinger, The dimensions of the grid box for 3EQM of aromatase receptor (Figure 3) were kept as: Inner box: X=12, Y=12, Z=13, Outer box: X=35, Y=30, Z=35, Grid center X=-16.817, Y=8.712, Z=-5.589.

Figure 3: Receptor structure aromatase enzyme with grid without parent molecule androstenedione
Validation of protein
The validation of aromatase receptor was performed by Ramchandran plot, to test the reliability and reproducibility of the docking protocols for the study (Figure 4).
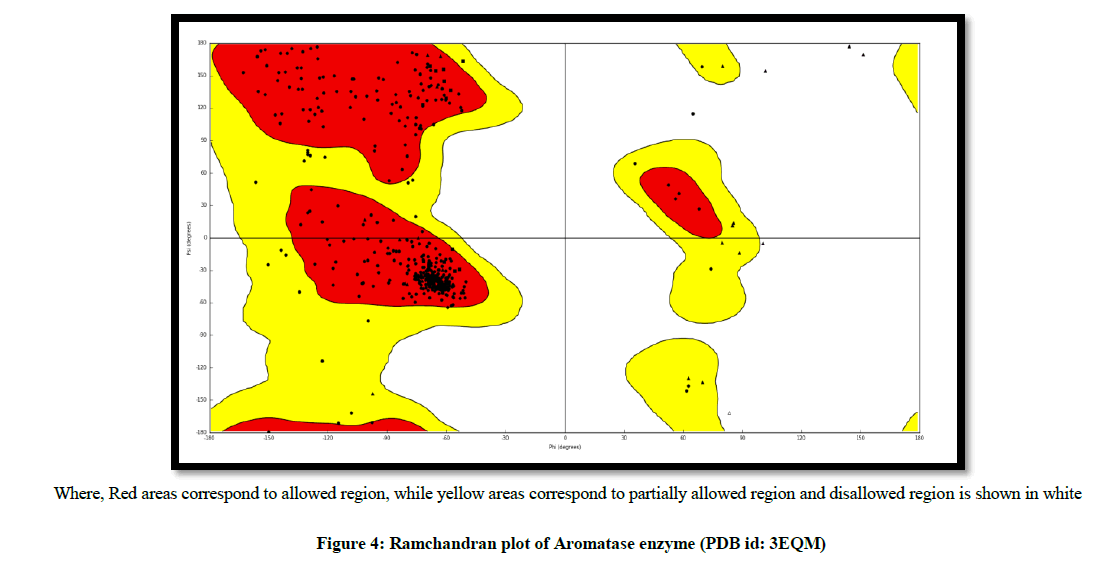
Where, Red areas correspond to allowed region, while yellow areas correspond to partially allowed region and disallowed region is shown in white
Figure 4: Ramchandran plot of Aromatase enzyme (PDB id: 3EQM)
Protein ligand docking
For the docking, twelve 3D flavonoid derivatives have been docked on aromatase enzyme with (PDB id: 3EQM). The ligand docking process helps to identify correct poses of ligands in the binding pocket of a protein and to predict the affinity between the ligand and protein. Firstly Ligand docking application was selected from Glide in the Application menu. Next to receptor grid, click Browse and choose generated receptor grid The LigPrep file of flavones was then browsed and entered into ligands to be docked. The ligand docking was carried out in the standard precision (SP) mode. The glide score, dock score and hydrogen bonding interaction of the synthesized compound were then compared with that of standard compound.
Materials and Methods
Experimental work
All chemicals were obtained from S.D. Fine chemicals, Loba Chemicals and Merck Pvt. Chemicals and solvents were purified by general laboratory techniques before use. All moisture free operations were performed in oven dried glassware’s and under nitrogen atmosphere. Melting points were determined by using Capillary tube method and are uncorrected. Ultraviolet spectra were obtained on Shimadzu 1700 UV-Visible spectrophotometer using methanol as a solvent. IR spectra (wave numbers in cm-1) were recorded on an IR-affinity-1S Shimadzu A219652, FT-IR spectrophotometer using potassium bromide discs. The progress of all the reactions were monitored by TLC on 2 cm × 5 cm pre-coated silica gel 60 F254 (Merck) plates of thickness of 0.25 mm. The chromatograms were visualized under UV 200-400 nm and/or exposure to iodine vapours.
Experimental procedure (Scheme 1)
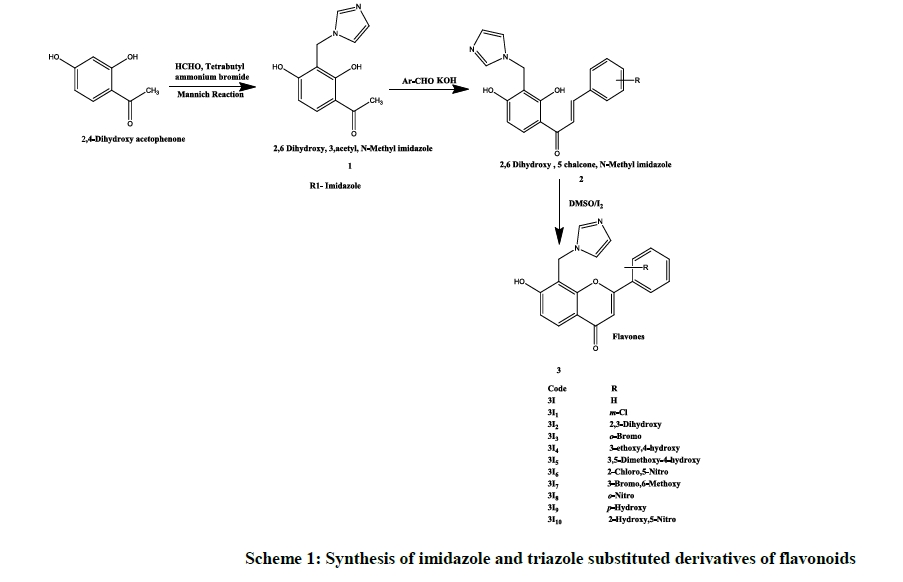
Scheme 1: Synthesis of imidazole and triazole substituted derivatives of flavonoids
2, 6-Dihydroxy, 5 acetyl, N-methyl imidazole (1)
2, 4-Dihydroxyacetophenone (0.002 mol) dissolved in chloroform (20 ml) and formaldehyde (0.004 mol), 1, 3-imidazole (0.004 mol), tetrabutyl ammoium bromide (0.002 mile) and 10 ml of distilled water was added to it. After stirring the content for 24 h chloroform layer was separated, washed with aqueous sodium bicarbonate (8% w/w) followed by washing with water. The organic layer then dried over anhydrous sodium sulphate, filtered and evaporated to dryness. The residue was crystallized by methanol to give TLC pure crystals [13,14].
2, 6 Dihydroxy, 5 chalcone, N-methyl imidazole (2)
A mixture containing compound 2 (5 mol) in ethanol (20 ml.), arylaldehyde (10 mol) and a solution of potassium hydroxide (3 g) in distilled water (5 ml) was stirred for 2 h at room temperature and then left overnight. It was poured into cold water and acidified with HCl, a solid mass, was separated out, filtered, washed with water, sodium bicarbonate solution (2% w/v in water) and again with water, purified with suitable solvent [13,14].
6-hydroxy-5-(N-methyl imidazole)-4-chromones (3I)
To a solution of compound 3 (200 mg) in dimethyl sulfoxide (5 ml), 2 crystals of iodine was added then this mixture was refluxed for 30 min., cooled to room temperature and poured into ice cold water. A solid mass was separated out, filtered, washed with water, sodium thiosulphate solution (2% w/v in water) and again with water after drying it was crystallized by suitable solvent. All the top rank flavonoids with highest docking score and binding energy were synthesized and characterized by UV and IR Spectroscopy. TLC was taken in Hexane: Ethyl acetate (9: 1) ratio (Table 1) [13,14].
| Compound | Structure | IUPAC Name | Characterization |
|---|---|---|---|
| 3I1 | 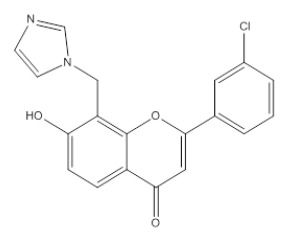 |
7 -Hydroxy, 8-Methyl imidazole, 2-(m-Chlorophenyl), 4-H, Chromene-4-One | λmax-235 nm IR (cm-1)- 1685 1593, 761, 1425, 1282, 1490, 852, 3150 |
| 3I2 | 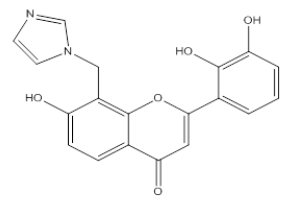 |
Hydroxy, 8-Methyl imidazole, 2-(2, 3-Dihydroxy, phenyl), 4-H, Chromene-4-One | λmax -237 nm IR (cm-1)-810, 1090, 1650, 1610, 1450, 3600, 1330, 1100 |
| 3I3 | 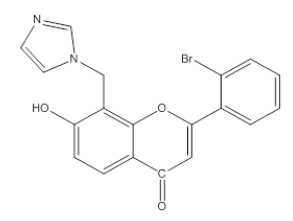 |
7-Hydroxy, 8-Methyl imidazole, 2-(O-Bromophenyl), 4-H, Chromene-4-One | λmax -239.8 nm IR (cm-1)-3100, 1690, 1600, 1430, 1487, 1068, 850, 1128, 1300, 758, 550 |
| 3I6 | 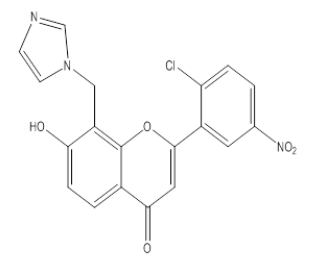 |
7 -Hydroxy, 8-Methyl imidazole, 2-(2-Chloro, 5-Nitrophenyl), 4-H, Chromene-4-One | λmax - 227 nm IR (cm-1)-1350, 810, 1100, 1090, 3090, 1650, 1610, 1450, 3600, 1510, 750 |
| 3I8 | 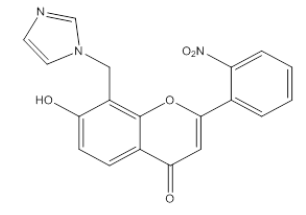 |
7 -Hydroxy, 8-Methyl imidazole, 2-(O-Nitrophenyl), 4-H, Chromene-4-One | λmax - 240 nm IR (cm-1)-3600, 1090, 3090, 1710, 1610, 1450, 750, 1500, 1250, 1150 |
| 3I10 | 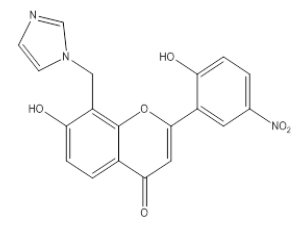 |
7 -Hydroxy, 8-Methyl imidazole, 2-(2-Hydroxy, 5-Nitro, phenyl), 4-H, Chromene-4-One | λmax - 232 nm IR (cm-1)-3100, 1650, 1610, 1430, 3600, 850, 1100, 1338, 1520 |
Table 1: Top rank Flavonoids with highest binding interaction with aromatase enzyme
Results and Discussion
Molecular docking was performed to elucidate the binding mode of aromatase and its inhibitors. To this end, a total of twelve novel flavonoids, were docked to the aromatase binding site. Scrodinger software was used to prepare the molecules and to show the interaction of the compound in binding pocket of an aromatase enzyme results indicated that the X-ray crystallography conformer was nearly identical to the docked conformer by giving the result of docking in the form of docking score, glide score and free binding energy. Binding pose with the lowest docked energy belonging to the top-ranked cluster was selected as the final model for post-docking analysis and has been synthesized by a modified scheme of synthesis and will be evaluated for the in vitro and in vivo Anti-Breast cancer activity. Results obtained from Scrodinger provided pertinent information on the binding orientation of ligand-receptor interactions. Docking results for the designed flavonoids along with reference inhibitor (Fadrazole) (Figure 5) are summarized (Table 2) for the comparative purpose. On the basis of docking score, Glide g-score we concluded that all the novel flavonoid are having higher docking and G-score than Fadrazole and compound namely 3I2 due to hydroxy substitution is more effective against 3EQM receptor with docking score (-8.603) and G score (-8.954) as compared to the earlier reported inhibitor (Fadrazole) with docking score (-5.536) and G score (-5.879) followed by 3I3, 3I13. The docking analysis revealed that the top-ranked flavonoids 3I2, 3I3, 3I13 forms H-bond interaction between the hydroxy group and LEU 372 and ALA 306 amino acid residue of 3EQM receptor. Pi-Pi interaction is formed between imidazole ring and PHE 221 and ARG 115 amino acid residue of 3EQM. Hydrophobic interactions with the amino acid residues TRP 224, VAL 370, LEU 372, VAL 369, MET 374, VAL 373, PHE 134, ILE 133, PHE 221, LEU 477, and ILE 305 were observed (Figure 6).
| Compound | Docking score (kcal/mole) | Glide score (kcal/mole) | Binding energy (kcal/mole) |
|---|---|---|---|
| 3I2 | -8.603 | -8.954 | 98.01 |
| 3I3 | -8.395 | -8.746 | 108.826 |
| 3I4 | -7.664 | -8.0817 | 99.536 |
| 3I5 | -6.588 | -6.931 | 97.059 |
| 3I6 | -8.063 | -8.41 | 132.051 |
| 3I7 | -7.163 | -7.509 | 158.015 |
| 3I8 | -7.355 | -7.711 | 110.251 |
| 3I9 | -7.283 | -7.634 | 118.849 |
| 3I12 | -7.429 | -7.784 | 124.308 |
| 3I13 | -8.032 | -8.378 | 87.821 |
| Fadrazole | -5.536 | -5.879 | 83.241 |
Table 2: Result of molecular docking simulation study
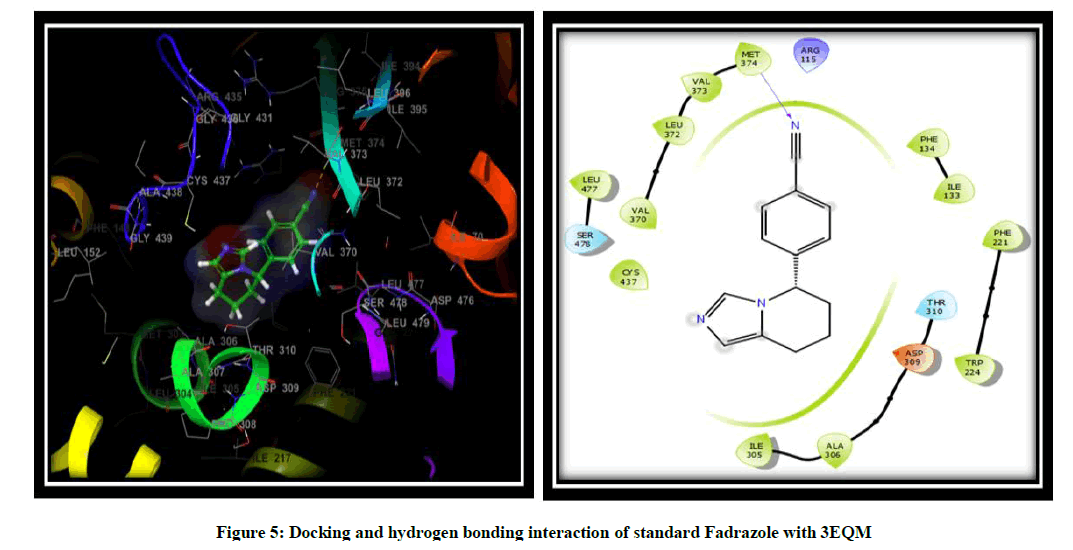
Figure 5: Docking and hydrogen bonding interaction of standard Fadrazole with 3EQM
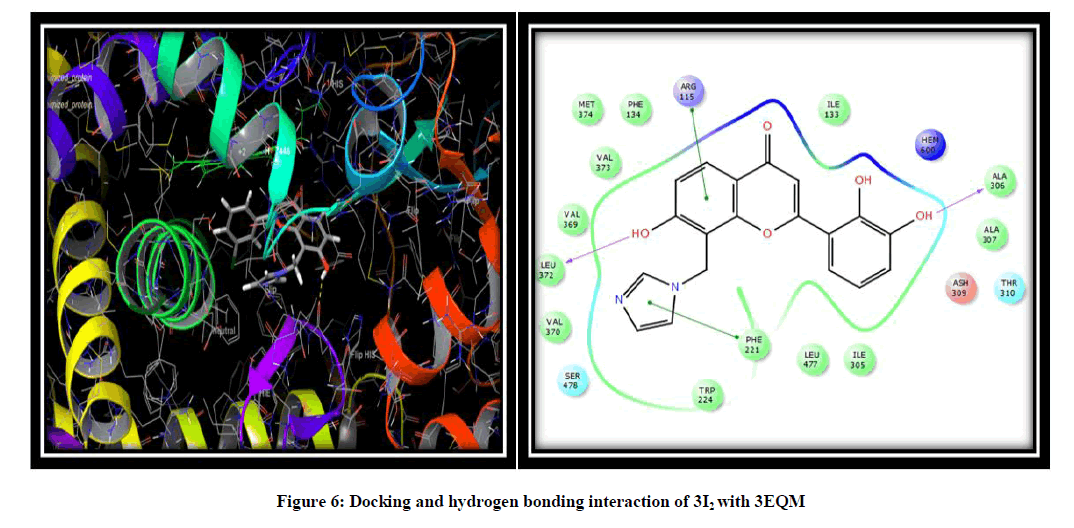
Figure 6: Docking and hydrogen bonding interaction of 3I2 with 3EQM
Conclusion
The results of the present study clearly demonstrated that the molecular docking studies of designed flavonoids with aromatase enzyme exhibited binding interactions and warranted further studies needed for the development of potent aromatase inhibitors for the treatment of Breast cancer. These results clearly indicated that flavonoids have shown more binding sites and interactions with aromatase enzyme compared to the reference. These docking studies are actually an added advantage to screen the aromatase inhibition. Flavonoids may serve as useful leads in the development of clinically useful aromatase inhibitors. Further, investigations on the above compound need in vitro and in vivo studies to develop potential chemical entities for the prevention and treatment of Breast cancer.
Acknowledgement
The authors are thankful to the Head, Department of Pharmaceutical Sciences, Rashtrasant Tukadoji Maharaj Nagpur University, Nagpur, for providing necessary facilities and other technical supports during the preparation of this article.
References
- Cancer Facts and Figures, American Cancer Society Center in Atlanta, Georgia, Inc., 2009, 1-72.
- Global Cancer Facts and Figures, American Cancer Society Center in Atlanta, Georgia, Inc., 2007, 1-59.
- A.K. Verma, R. Pratap, Tetrahedron., 2012, 30, 1-16.
- T. Lijun, Z. Shufen, Y. Jinzong, G. Wentao, C. Jian, Z. Tianyu, Molecules., 2004, 9, 842-848.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, Natl. Cancer Inst., 1990, 82(13), 1107-1112.
- D. Ghosh, J. Griswold, M. Erman, W. Pangborn, Nature., 2009, 457, 219-23.
- S. Costanzi, I.G. Tikhonova, T.K. Harden, K.A. Jacobson, J. Computer-Aided Molec. Design., 2009, 23, 747-754.
- S. Vilar, S. Costanzi, Methods in Molec. Biol., 2012, 914, 271-284.
- L.M. Demers, Breast Cancer Res. Treat., 1994, 30, 95-102.
- M. Awasthia , S. Singha , V.P. Pandeya, U.N. Dwivedia, J. Biomolec. Struc. Dynam., 2014.
- H.A. Zhu, X.H. Liu, H.F. Liu, S. Xu, A.S. Bao, P.S. Bhadury, J.X. Liu, X.B. Qi, Bioorg. Med. Chem.,2010, 20, 4163-4167.
- N. Suvannang, C. Nantasenamat, C. Isarankura-Na-Ayudhya, V. Prachayasittikul, Molecules., 2011,16, 3597-3617.
- A. Husain, S. Sharma, Acta Poloniae Pharmaceutica-Drug Res., 2010, 67, 3, 261-266.
- A. Husain, M. Rashid, D. Kumar, Acta Poloniae Pharmaceutica-Drug Res., 2013, 67, 3, 443-449.



