Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 3
Nephroprotective Potentials of Green Tea through MPTP in Gentamicin Induced Nephrotoxicity in Rats
Gurfateh Singh1*, Hardeep Kaur1, Deepika Bhatia2 and Monisha Bansal1
1University School of Pharmaceutical Sciences, Rayat Bahra University, Kharar, Mohali, Punjab, India
2University Institute of Pharmaceutical Sciences, Chandigarh University, Gharaun, Mohali, Punjab, India
- *Corresponding Author:
- Gurfateh Singh
University School of Pharmaceutical Sciences
Rayat Bahra University
Kharar, Mohali, Punjab, India
Abstract
Purpose: To investigate the nephroprotective potentials Green tea extracts dose dependently in gentamicin induced experimental nephrotoxicity via Mitochondrial Permeability Transition Pores (MPTP).
Methods: Six groups of rats employed in present study each group comprises 5 animals. Experimental nephrotoxicity induced by administration of gentamicin (80 mg/kg body weight/day, i.p) for six consecutive days. The thiobarbituric acid reactive substances, superoxide dismutase and glutathione levels in the kidney tissues measured for oxidative stress. Nephrotoxicity evaluated by serum creatinine, Blood Urea Nitrogen (BUN) and histopathological examination.
Results: Gentamicin induced nephrotoxicity characterized by a significant increase in BUN, and serum creatinine levels. Moreover oxidative stress was noticed in renal tissue as evidenced by a significant decrease in glutathione level, superoxide dismutase also a significant increase in malondialdehyde levels in gentamicin & atractyloside treated rats. Pre-administration of green tea extract (100 or 200 mg/kg once daily, p.o) for 15 days restored renal functions and attenuated oxidative stress.
Conclusion: Green tea extract ameliorates gentamicin induced nephrotoxicity and atractyloside induced opening of MPTP leading oxidative damage by scavenging oxygen free radicals, decreasing lipid peroxidation and improving intracellular antioxidant defense and may closing of MPTP.
Keywords
Gentamicin, Atractyloside, Nephrotoxicity
Introduction
Nephrotoxicity refers to dangerous kidney problem that develop when toxins accumulate in the kidneys. Toxins and unneeded fluids are usually excreted through urine, but when they start reaching excessive levels, they eventually lead to nephrotoxicity and cause a variety of other symptoms of kidney trouble [1]. Chronic Kidney Disease (CKD), which is characterized by a chronic reduction in the Glomerular Filtration Rate (GFR) and the presence of proteinuria or albuminuria, is recognized as an independent risk factor for both End-Stage Renal Disease (ESRD) and cardiovascular disease, leading to a decrease in quality of life and an increased risk of mortality [2]. Acute Kidney Injury (AKI) is common in the setting of critical illness and is associated with a high risk of death [3]. In addition, AKI can directly cause ESRD and can increase the risks of the development of incident CKD and the worsening of underlying CKD [4]. There are several agents induced nephrotoxicity like heavy metals, anti-neoplastic agents, antimicrobial agents, aminoglycosides and radio contrast agents.
Mitochondria are playing a vital role in apoptosis and necrotic cell death [5]. It has been noted that gentamicin produced apoptosis, [6] necrosis [7] of tubular epithelial cells in vivo and also in cultured cells [8]. It has been noted that gentamicin released into the cytosol from where it acts on mitochondria and activated the mitochondrial pathway of apoptosis, produced oxidative stress, and reduced the ATP synthesis [9,10]. Further apoptosis activated the caspase-3 and caspase-7 which leads to DNA fragmentation due to blabbing of membrane and shrinkage of cell. The opening of Mitochondrial Permeability Transition Pores (MPTP) due to ROS and cytokines was first described as necrotic cell death. MPTP opening leads to more oxidative stress and renal toxicity. The opening of MPTP leads to oxidative stress. MPTP can be triggered by multiple factors such as elevated mitochondrial Ca2+ increased ROS and a variety of lipid metabolites. Persistent MPT leads to the loss of mitochondrial membrane potential, mitochondrial swelling, outer membrane rupture and consequent release of apoptogenic factors [11,12].
The health benefits of consuming green tea (GT) has been reported, including the prevention of Cancer [13], Cardiovascular diseases [14], anti-inflammatory [15], antiarthritic [16], antibacterial [17], antiangiogenic [18], antiviral [19], neuroprotective [20] and cholesterol-lowering effects. GT inhibit DNA damage [21], decrease ROS generation, formation of peroxynitrates, expression of Peroxisome Proliferator-activated Receptor Gamma (PPAR-γ), Interleukins (ILs) formation and Tumor Necrosis Factor Alpha (TNF-α) [15]. GT suppressed the phosphorylation of Epidermal Growth Factor Receptor (EGFR) [22] and regulate JAK/STAT, MAPK, PI3K/AKT survival pathways. Moreover GT increased ATP generation and suppressed oncogenic transcription factors [23] angiogenesis by suppressing VEGF activity, VE-cadherin phosphorylation and matrix metalloproteinase activity and activated the killer Caspases [24].
Atractyloside (ATR) is found in many Asteraceae plants. It is an extremely toxic glucoside that is obtained from the Mediterranean thistle Atractylis gummifera. It inhibits oxidative phosphorylation by blocking the transfer of adenosine nucleotides through the mitochondrial membrane by the transport protein ATP-ADP translocase. The biological potential of Green Tea Extract (GTE) by attenuating MPTP is not documented yet. MPTP activation has been reported to be activated by high oxidative stress and lead to nephrotoxicity. Further, over activation of Ca2+, ROS was shown to opening of MPTP in gentamicin induced nephrotoxicity [11,12].
Therefore, the present study has been designed to investigate the nephroprotective potential of GTE on MPTP in gentamicin induced nephrotoxicity in rats.
Materials and Methods
Drugs and chemicals
All the drug solutions were freshly prepared before use. Gentamicin (GM) was purchased from Kumar Brothers Pvt. Ltd., PGI, Chandigarh. Green tea extract was obtained from Omkar Herbals Pvt. Ltd, Indore, India as a gift sample. Atractyloside was purchased from Ausmasco Pvt. Ltd, China. Diagnostic kits were purchased from Reckon Diagnostics Pvt. Ltd. Vadodara, India. All other chemicals/reagents were analytical grade and obtained from the central store of our Institute.
Experimental protocol
Six groups were employed in the present study and each group comprised of five animals. The animals were housed in a room temperature, humidity controlled room and a 12 h light-dark cycle. Rats were allowed free access to purified water and standard pellet diet. The institutional Animal Ethics Committee/CPCSEA approved experimental protocol.
GROUP-1 (Normal control): Rats were treated with normal saline for 15 days.
GROUP-2 (Atractyloside control): Animals were administered with atractyloside (5 mg/kg p.o) for 15 days and sacrificed on 16 days.
GROUP-3 (Gentamicin treated control): Animals were treated with gentamicin (80 mg/kg i.p) for 6 consecutive days started from day 10th to 15th days of experimental protocol.
GROUP-4 (Gentamicin pre-treated GTE-100 mg): The administration of GTE (100 mg/kg p.o) for 15 days in gentamicin treated rats.
GROUP-5 (Gentamicin pre-treated GTE-200 mg): The administration of GTE (200 mg/kg p.o) for 15 days in gentamicin treated rats.
GROUP-6 (Gentamicin pre-treated GTE-200 mg & atractyloside): The administration of GTE (200 mg/kg p.o) and atractyloside (5 mg/kg p.o) for 15 days in GM treated Rats.
Preparation of renal homogenate
The tissue from isolated fresh kidney of experimental rats was minced and homogenized in 0.1 M ice cold phosphate buffer (pH 7.4) using a homogenizer. The clear supernatant of the homogenate was used to estimate the Thiobarbituric Acid Reactive Substances (TBARS), Superoxide Dismutase (SOD) and Glutathione (GSH) levels in the tissue.
TBARS
The tissue lipid peroxidation reaction was assessed by estimating TBARS using method of Wills [25] with some modifications. Briefly, 500 μl homogenate was added to equal amount of the same buffer, and incubated for 2 h at 37°C. After incubation, 1 ml of 10% Trichloroacetic Acid (TCA) was added to the mixture and centrifuged at 3000 rpm for 10 min. The supernant (1 ml) was added to 1 ml of 0.67% TBA, solution and heat for 15 min on water bath. The samples were cooled and 1 ml of distilled water was added. The color intensity was determined at 532 nm. TBA reacting compound was expressed as nM of Malonaldehyde (MDA)/mg of tissue [26].
Estimation of GSH
The reduced GSH level was assessed by the method of Ellman. Briefly, the tissue homogenate in 0.1 M phosphate buffer pH 7.4 was mixed with equal volume of 20% TCA containing 1 mM EDTA to precipitate the tissue proteins. The mixture was allowed to stand for 5 min prior to centrifugation for 10 min at 200 rpm. The supernatant (200 μl) was then transferred to a new set of test tubes and added 1.8 ml of the Ellman's reagent 5,5'-Dithio-bis-2-nitrobenzoic acid (DTNB) (0.1 mM) was prepared in 0.3 M phosphate buffer with 1% of sodium citrate solution. Then, all the test tubes make up to 2 ml volume. The absorbance of the solution was measured at 412 nm against blank and the amount of reduced GSH was expressed as nM/mg of tissue [27].
Estimation of SOD
SOD activity was measured according to a method by Misra and Fridovich [28], by following spectrophotometrically the auto-oxidation of epinephrine at pH 10.4. In this method, supernatant of the tissue was mixed with 0.8 ml of 50 mM glycine buffer, pH 10.4, and the reaction was started by the addition of 0.02 ml (−)-epinephrine. After 5 min, the absorbance was measured at 480 nm. The activity of SOD was expressed as percent activity of vehicle-treated control [29].
Blood collection and biochemical assays’
Blood samples were withdrawn from the retro-orbital of each animal using a glass capillary tube. The blood samples allowed to coagulate and then centrifuged at 3,000 rpm for 20 min. The separated sera were used for the estimation of serum activities.
Assessment of gentamicin induced nephrotoxicity
Nephrotoxicity was determined by estimation the level of creatinine, Blood Urea Nitrogen (BUN) in the blood serum using commercially available kits. Values were expressed in mg/dl. The serum/urine creatinine concentration was estimated by alkaline picrate method using the commercially available kit.
Histopathological examination
The kidneys were sectioned longitudinally into two halves and were kept in 10% neutral formalin solution. One kidney were processed and embedded in paraffin wax and sections were taken using a microtome. Sections of 5 μm thickness cut and stained with hematoxyline and eosine.
Paraffin wax removed by warming the glass slide gently, observed under a computerized light microscope. The melted wax removed with Xylene then slide washed with absolute alcohol and water to hydrate the section then stained with Hematoxylin-Eosine. In brief the micro photo graphs of the kidney sections were captured and necrotic areas were identified and calculated.
Statistical analysis
The observations were statistically analyzed with the help of InStat. All of the results were expressed as the mean ± SEM. Results were analyzed using one-way Analysis OF Variance (ANOVA), followed by Tukey’s multiple comparison test and p <0.05 was considered to be statistically significant.
Results
Rats fed with gentamicin (80 mg/kg/day; i.p.) for 6 days produced nephrotoxicity when compared with normal rats. The nephrotic injury parameters like creatinine and BUN were noted to be increased significantly in gentamicin treated rats when compared with normal rats. Moreover the lipid peroxidation measured in terms of reduced NBT were noted to be increased significantly but the reduced form of GSH and SOD activities were found to be decreased in gentamicin treated rats. The pathological changes of kidney cells were also observed in treated rats by histopathological evaluation. Gentamicin administration did not produce any mortality of rats in the present study.
Effect of green tea treatment on kidney functions of rats
Gentamicin produced a significant elevation in the serum creatinine level from 0.19 ±0.02 to 2.82 ± 0.48 mg/dl. In addition, the level of serum creatinine significantly increased but not markedly increased was observed in Atractyloside treated group due to the opening of mitochondrial permeability transition pore. On the other hand simultaneous pre-administration of GTE (100 mg/kg-low dose and 200 mg/kg-high dose) for 15 days with GM showed significant reduction of serum creatinine levels. Treatment with GTE (High dose) and atractyloside (5 mg/kg) for 15 days significantly abolished the observed nephroprotective potential of GTE characterized by increased level of serum creatinine (Figure 1).
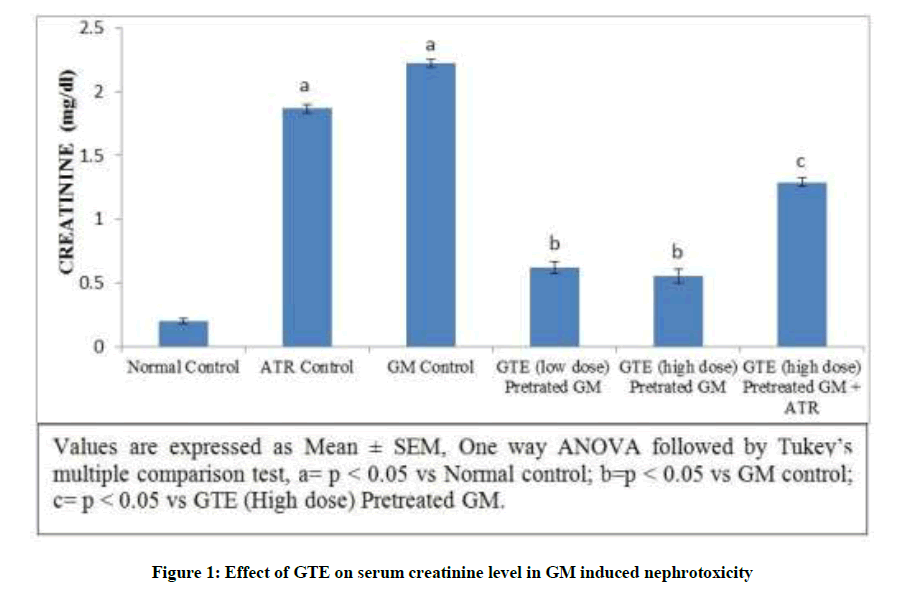
Figure 1: Effect of GTE on serum creatinine level in GM induced nephrotoxicity
Effect of GTE on serum BUN in GM induced nephrotoxicity: Gentamicin produced a significant elevation in the BUN level from 22.7239 ± 1.03 to 59.0577 ± 3.78 mg/dl. In addition, the level of BUN significantly increased but not markedly increased was observed in Atractyloside control due to opening of mitochondrial permeability transition pore. On the other hand simultaneous pre-administration of GTE (low & high dose) for 15 days with GM showed significant reduction of BUN level dose dependently. Treatment with GTE (High dose) and Atractyloside (5 mg/kg) for 15 days significantly abolished the observed nephroprotective effect of GTE when compared with GTE pretreated GM rats (Figure 2).
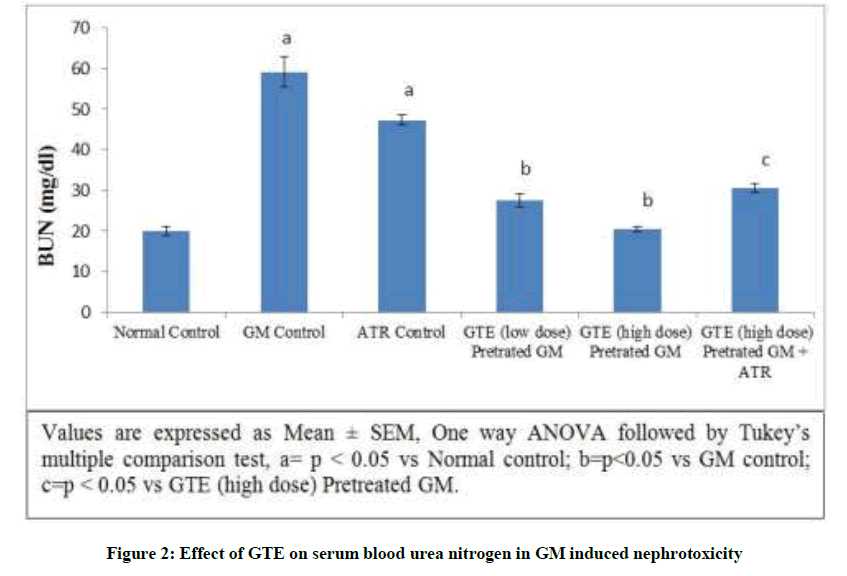
Figure 2: Effect of GTE on serum blood urea nitrogen in GM induced nephrotoxicity
Potentials of GTE on renal oxidative stress biomarkers
Effect of GTE on TBARS in GM induced nephrotoxicity: Gentamicin produced a significant elevation in the TBARS level from 0.7808 ± 0.0280 to 3.6352 ± 0.1269 nmol/mg of tissue. In addition, the level of TBARS significantly increased observed in Atractyloside control group. On the other hand simultaneous pre-administration of GTE (Low dose & high dose) for 15 days with GM showed significant reduction of TBARS level dose dependently. Treatment with GTE (high dose) and atractyloside (5 mg/kg) for 15 days significantly abolished the nephroprotective effect of GTE (Figure 3).
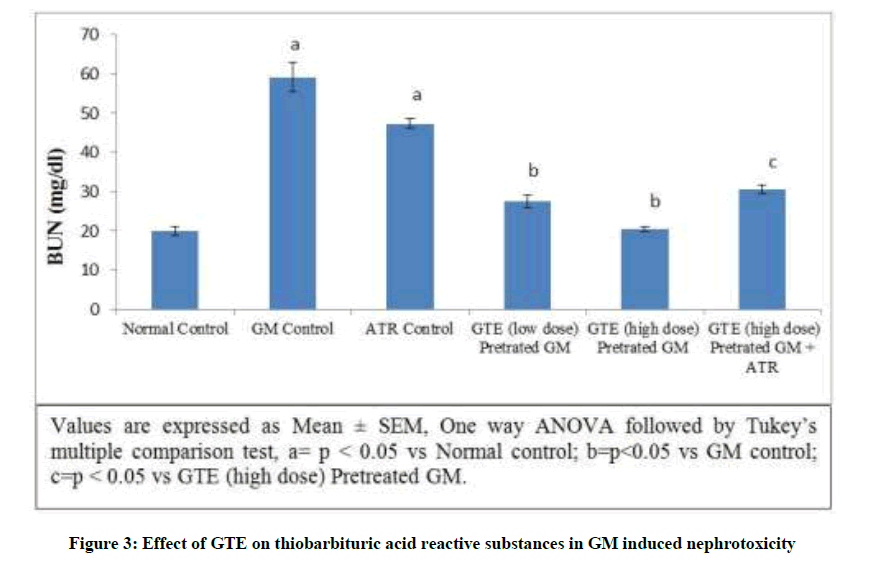
Figure 3: Effect of GTE on thiobarbituric acid reactive substances in GM induced nephrotoxicity
Effect of GTE on SOD activity in GM induced nephrotoxicity: Gentamicin produced a significant reduction in the SOD level. In addition, the level of SOD significantly decreased observed in Atractyloside control group. Pre-administration of GTE (Low dose & high dose) significant elevated SOD level. Treatment with GTE (High dose) and atractyloside (5 mg/kg) significantly abolished the nephroprotective effect of GTE (Figure 4).
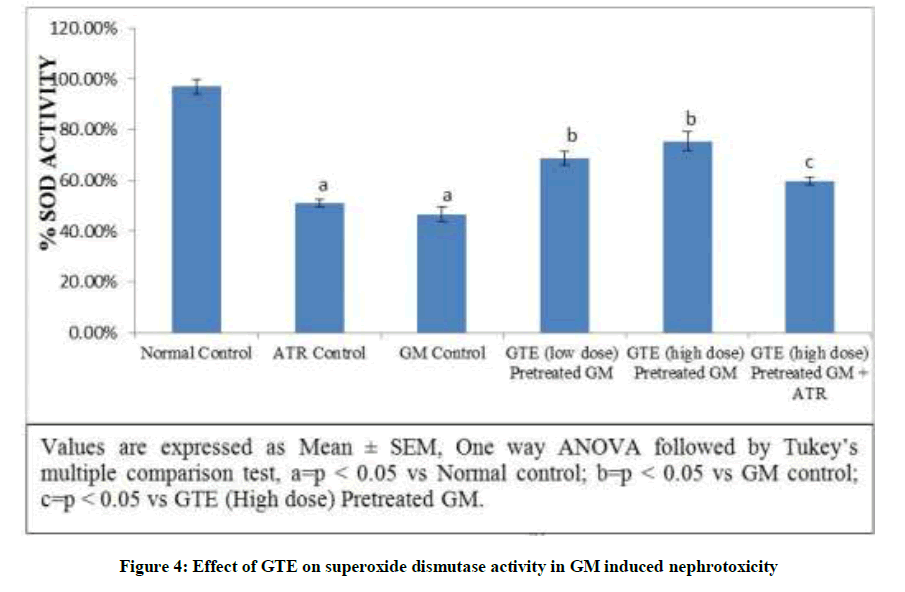
Figure 4: Effect of GTE on superoxide dismutase activity in GM induced nephrotoxicity
Effect of GTE on GSH level in GM induced nephrotoxicity: GSH has a very important role in protecting against oxygen free radical damage by providing reducing equivalents for several enzymes. Gentamicin produced a significant reduction in the GSH level. In addition, the level of GSH significantly decreased in Atractyloside group. On the other hand simultaneous pre-administration of GTE (Low & high dose) for 15 days with GM showed significant elevation of GSH level dose dependently. Treatment with GTE (High dose) and Atractyloside for 15 days significantly abolished the nephroprotective effect of GTE (Figure 5).
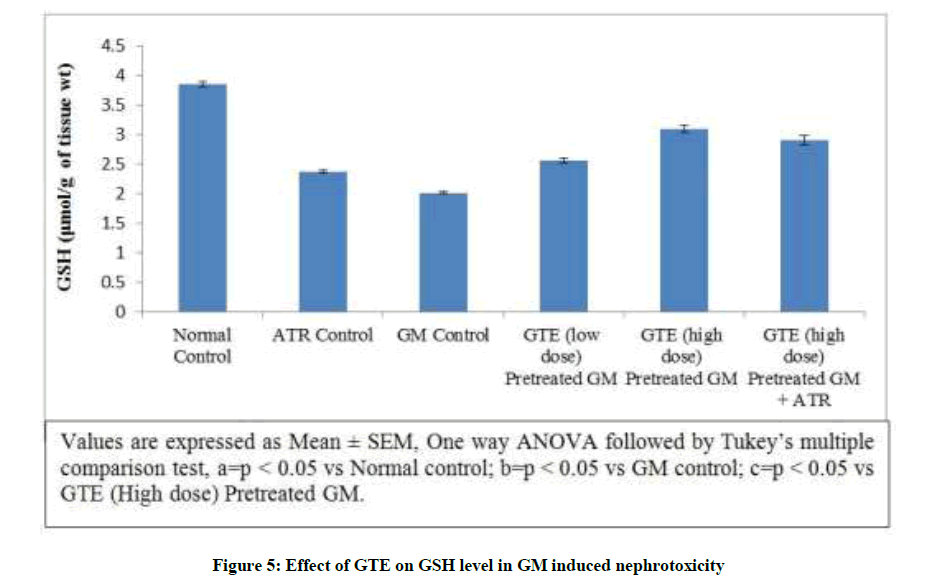
Figure 5: Effect of GTE on GSH level in GM induced nephrotoxicity
Effect of GTE on histopathological studies (100X) in GM induced nephrotoxicity: The histological changes of kidney features were observed in experimental protocol. The kidney cells in control rats were looking normal architecture with well-defined tubules and glomeruli. ATR control group indicated swelling, inflammation and acute damage of tubule and glomeruli. GM caused severe degeneration of tubular cells, cloudy cell swelling of tubule, glomeruli, damage of brush border line and inflammation. GTE (Low & high dose) supplementation reduced the cellular toxicity and reduced glomeruli and tubular damage caused by GM and restored a near normal morphological features were observed in group receiving GTE pretreated GM. Quantification of kidney sections revealed that GTE significantly reduced the necrotic areas. Treatment with GTE (High dose) and atractyloside significantly abolished the nephroprotective effect of GTE in GM treated rats when compared with EGCG pretreated GM (Figure 6).
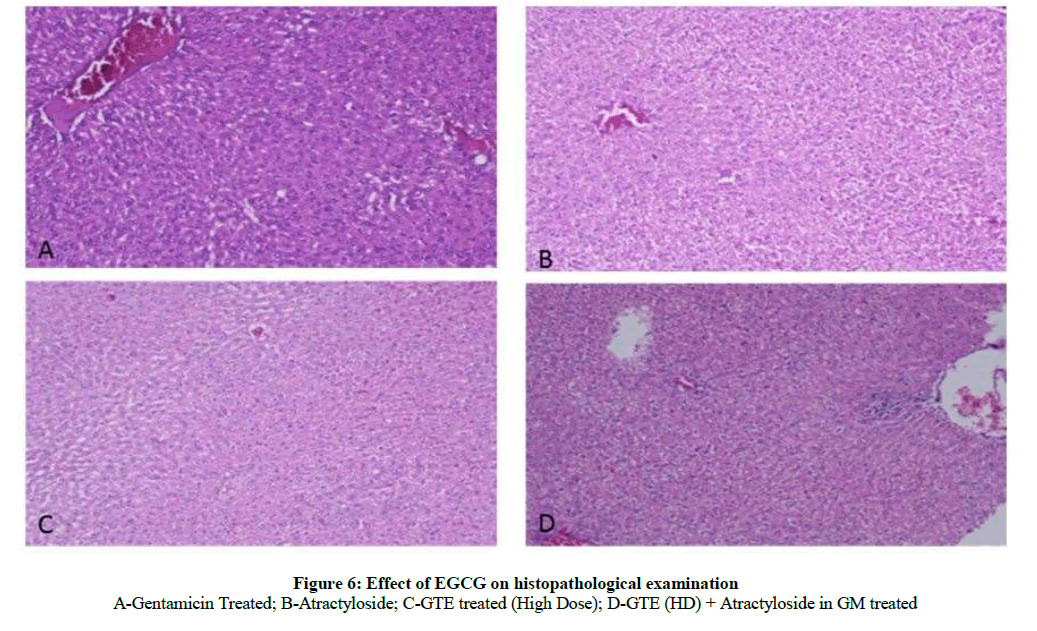
A-Gentamicin Treated; B-Atractyloside; C-GTE treated (High Dose); D-GTE (HD) + Atractyloside in GM treated
Figure 6: Effect of EGCG on histopathological examination
Discussion
In spite of undesirable Gentamicin-induced nephrotoxicity, this antibiotic still constitute the only effective therapeutic alternative against microorganisms insensitive to other antibiotics. Moreover, gentamicin-induced nephrotoxicity has been widely used as an animal model to study acute kidney failure in experimental research [30]. The finding of present study revealed that the GTE has therapeutic potential to ameliorate gentamicin induced kidney damage in rats.
Gentamicin is reported to produce kidney injury at the dose 40 mg/kg/day and 80 mg/kg/day is the well reported model for screening of nephroprotective agents against drug induced kidney injury. The various mechanisms such as induction of ROS, apoptosis, necrosis, elevation of endothelin I and increased in monocyte/macrophages infiltration involved in GM induced nephrotoxicity. It has been noted that elevation of NF- κB [31], Trans-forming Growth Factor-beta (TGF-β) [32] and iNOS production contributed to nephrotoxicity in rats [33]. Moreover, activation of cytochrome C, caspases, oxidative stress and increased calcium concentration is noted to induce mitochondrial dysfunction through activation of Mitochondrial Permeability Transition Pore (MPTP) and Adenosine Triphosphate (ATP) depletion in GM induced nephrotoxicity.
The magnitude of serum creatinine and blood urea nitrogen elevation has been shown to correlate strongly with renal impairment. The serum creatinine and blood urea nitrogen is an important biomarker which increases in gentamicin induced nephrotoxicity. The increase in creatinine and blood urea nitrogen is documented to be an index of kidney injury.
In the present study GM 80 mg/kg body weight caused marked kidney damage as observed increase level of serum creatinine and BUN. Gentamicin at the dose 40 mg/kg and 80 mg/kg are reported to cause increase level of serum creatinine and BUN in rodents. Serum creatinine concentration is a more potent indicator than the urea in the first phases of kidney disease. Furthermore, urea concentrations begin to increase only after parenchymal injury [34]. In our study, administration of gentamicin (80 mg/kg/day, i.p) led to significant increase of serum creatinine and blood urea nitrogen as it was previously reported by others [35,36].
On the other hand GTE (100 mg/kg and 200 mg/kg) significantly reduced the level of BUN and serum creatinine. BUN and creatinine are the metabolic enzymes and significantly increased level of above enzymes due to tubular damage by ROS generation [37], iNOS [38], TNF-α, NF kappa B, DNA damage, activation of cytosolic calcium, release of cytochrome c and deactivation of ATP due to MPTP opening. Therefore in our study GTE may have beneficial potentials to block the activation of ROS generation, iNOS, MAPK, TNF-α, NF kappa B, DNA damage, activation of cytosolic calcium, deactivation of ATP, release of cytochrome c and deactivation of ATP leads to MPTP changes result increased level of serum creatinine and BUN upon injury. GTE significantly decreased the level of BUN dose dependently but not creatinine. However GTE significantly blocks generation of ROS [21], nitric oxide, NF kappa B, interleukins formation, TNF alpha and increased ATP generation [15]. It has been observed that GTE up regulate ATP synthesis leading to nephroprotection responsible for increase the above mechanism in serum upon GM induced kidney damage.
GM induced kidney injury via cytochrome c release, activation of caspases (3 and 7) and increased expression of iNOS [39], leading to oxidative stress and increase release of pro inflammatory mediators like interleukins, TNF-α and interferon’s. These consequences lead to mitochondrial dysfunction which characterized by increase in Ca++ and leads to ATP depletion [40]. However in present study GTE (Low dose and high dose) treatment showed significant decreased the level of TBARS and increased the level of SOD and GSH. Moreover GTE showed significant decreased he level of TBARS and significant increased the level of GSH dose dependently. Hence GTE has potential to attenuated cellular events responsible for mitochondrial dysfunction and thereby preventing MPTP opening. This content is supported by earlier study mentioning that GTE at the dose of 100 mg/kg decreased oxidative stress, i.e., TBARS, SOD and GSH in rats [41].
The protective effects of GTE (High dose) in ameliorating GM induced nephrotic changes by preventing the MPTP opening is may confirmed by the treatment of atractyloside (selective MPTP opener) in present study since atractyloside alone is unable to cause marked nephrotic injury but only caused inflammation as in the form of significant increase in serum bio chemicals and tissue bio chemicals as compared to normal control. In the present study these results are found to be significantly low as compared to gentamicin control.
Gentamicin alters glomerular filtration rate because of mesangial cell contraction, loss of glomerular filtration barrier selectively due to the neutralization of its negative charges, mesangial cell proliferation, and apoptosis [31]. In the present study, the gentamicin-treated rats showed severe proximal tubular necrosis, cellular degeneration, degeneration of brush border line, swelling of tubular cells and inflammation in cell of rat kidney. Gentamicin induced these histopathological changes which are supported by earlier literature [31]. Tubular necrosis, degeneration of brush border line and cloudy inflammation was inhibited in GTE (Low and high dose) gentamicin treated rats in comparison with gentamicin treated animals. However GTE (High dose) significantly restored normal architecture of kidney and ameliorated gentamicin induced histopathological changes and this protection was founded to be attenuated on treatment with atractyloside due to opening of MPTP.
GTE has various pharmacological activities like the prevention of cancer [13], cardiovascular diseases [14], Inflammation [15], arthritic [16], bacterial infections [17], angiogenic effect [18], viral infections [19], moreover it shown neuroprotective [20] and cholesterol-lowering effects also. Hence, the protective effect of GTE was achieved due to its biological potential may be by closing of MPTP which was further may confirmed by its attenuation using Atractyloside an MPTP opener in the present study.
Conclusion
It may be concluded that the high degree of oxidative stress produced by the administration of gentamicin in rats and consequent opening of MPTP may be responsible for nephrotoxic effects of gentamicin and treatment of GTE (100 and 200 mg/kg, p.o) shown nephroprotective potentials via may closing of MPTP. The novel finding may concluded not to use Atractyloside in higher dosage in future biological studies due to inflammation of kidney and nephrotoxic effects. Isolation and characterization of active phytoconstituents from GTE can be done and their biological potentials in amelioration of renal dysfunction can be documented. This study provide for the first time scientific rational of using GTE in prevention or cure of renal injury caused due to drug or chemicals on chronic usage by closing of MPTP. However GTE which is a potent antioxidant prevent the opening of MPTP and thereby arrested mitochondrial dysfunctioning upon gentamicin exposure.
Acknowledgement
We wish to express our gratefulness to Dr. S.L. Harikumar (Honorable Dean USPS), Dr. Raj Singh (Honorable Vice Chancellor) Sr. Gurvinder Singh Bahra Ji (Honorable Chancellor), of Rayat-Bahra University Mohali (Punjab) for their praiseworthy inspiration, platform and constant support for the completion of this study.
References
- N. Kennedy, Ezine Articles., 2010.
- A. Levin, P.E. Stevens, Ann. Intern. Med.,2013, 158(11), 825-830.
- S. Uchino, J.A. Kellum, R. Bellomo, J. Am. Med. Assoc., 2005, 294(7), 813-818.
- L.S. Chawla, P.L. Kimmel, Kidney Int., 2012, 82(5), 516-524.
- C.L. Yang, X.H. Du, Y.X. Han, Ren. Fail., 1995, 17, 21-26.
- S. Li, K.K. Nagothu, V. Desai, T. Lee, W. Branham, C. Moland, J.K. Megyesi, M.D. Crew, D. Portilla, Kidney Int., 2009, 76, 1049-1062.
- J.R. Edwards, E.A. Diamantakos, J.D. Peuler, P.C. Lamar, W.C. Prozialeck, BMC Physiol., 2007, 7, 1.
- E.A. Pessoa, M.B. Convento, R.G. Silva, A.S. Oliveira, F.T. Borges, N. Schor, Braz. J. Med. Biol. Res., 2009, 42, 614-620.
- C.F. Simmons, R.T. Bogusky, H.D. Humes, J. Pharmacol. Exp. Ther., 1980, 214, 709-715.
- A.I. Morales, D. Detaille, M. Prieto, A. Puente, E. Briones, M. Arevalo, X. Leverve, J.M. Lopez-Novoa, M.Y. El-Mir, Kidney Int., 2010, 77, 861-869.
- C.P. Baines, R.A. Kaiser, N.H. Purcell, N.S. Blair, H. Osinska, M.A. Hambleton, E.W. Brunskill, M.R. Sayen, R.A. Gottlieb, G.W. Dorn, J. Robbins, J.D. Molkentin, Nature., 2005, 434, 658-662.
- A. Rasola, M. Sciacovelli, B. Pantic, P. Bernardi, FEBS Lett., 2010, 584, 1989-1996.
- G.P. Yu, C.C. Hsieh, Cancer Causes Contrl., 1995, 6, 532-538.
- P. Bhardwaj, D. Khanna, Chin. J. Nat. Med., 2013, 11, 345-53.
- H.S. Oz, T. Chen, W.J. deVilliers, Front Immunol., 2013, 4, 132.
- S. Riegsecker, D. Wiczynski, M.J. Kaplan, S. Ahmed, Life Sci., 2013, 93(8), 307-12.
- A. Sharma, S. Gupta, I.P. Sarethy, S. Dang, R. Gabrani, Food Chem., 2012, 135, 672-675.
- M. Demeule, J. Michaud-Levesque, B. Annabi, D. Gingras, D. Boivin, J. Jodoin, S. Lamy, Y. Bertrand, R. Béliveau, Curr. Med. Chem. Anticancer Agents., 2002, 2(4), 441-63.
- H.S. Kim, V. Montana, J. Biol. Chem., 2013, 288, 22693-22705.
- A.L. Lardner, Nutr. Neurosci., 2014, 17(4), 145-55.
- Y. Saffari, S.M.H. Sadrzadeh, Life Sci.,2004, 74, 1513-1518.
- S. Song K. Krishnan, K. Liu, R.S. Bresalier, Clin. Cancer Res., 2009, 15, 622-631.
- B.N. Singh, S. Shankar, R.K. Srivastava, Biochem. Pharmacol., 2011, 82, 1807-1821.
- W.W. Ting, M.S. Stone, K.C. Madison, K. Kurtz, Arch. Dermatol., 2003, 139, 903-906.
- E.D. Wills, Biochem. J.,1966, 99, 667.
- S.K. Sharma, N. Goyal, Zhong Xi Yi Jie He Xue Bao., 2012, 10, 555-560.
- S. Akbulut, H. Elbe, C. Eris, Z. Dogan, G. Toprak, E. Otan, E. Erdemli, Y. Turkoz, World J. Gastroenterol., 2014, 20, 10158-10165.
- H.P. Misra, I. Fridovich, J. Biol. Chem., 1972, 247, 3170-3175.
- D.K. Dhull, A. Jindal, R.K. Dhull, S. Aggrwal, D. Bhateja, S.V. Paddi, J. Mol. Neurosci., 2011, 1-9.
- E.O. Farombi, M. Ekor, Food Chem. Toxicol., 2006, 44, 1443-1448.
- J.M. Lopez-Novoa, Y. Quiros, L. Vicente, A.I. Morales, F.J. Lopez-Hernandez, Kidney Int., 2011, 79, 33-45.
- G. Bledsoe, S. Crickman, J. Maoetal, Nephrology Dialysis Transplantation., 2006, 21, 624-633.
- J.S. Christo, A.M. Rodrigues, M.G. Mouro, M.A. Cenedeze, J. Simes, N. Schor, Nitric Oxide, 2011, 24, 77-83.
- D.N. Gilbert, C.A. Wood, P.W. Kohlepp, D.C. Houghton, H.C. Finkbeiner, J. Lindsley, J. Infect. Dis., 1989, 159, 945-953
- P.D. Walker, Y. Barri, S.V. Shah, Ren Fail.,1999, 21, 433-442.
- B. Nitha, K.K. Janardhanan, Food Chem. Toxicol., 2008, 46, 3193-3199.
- A.I. Morales, D. Detaille, M. Prieto, A. Puente, E. Briones, M. Arevalo, X. Leverve, J.M. Lopez-Novoa, M.Y. El-Mir, Kidney Int., 2010, 77, 861-869.
- E. Ozbek, M. Cekmen, Y.O. Ilbey, A. Simsek, E.C. Polat, A. Somay, Ren Fail., 2009, 31, 382-392.
- H. Servais, A. Ortiz, O. Devuyst, S. Denamur, P.M. Tulkens, M.P. Mingeot-Leclercq, Apoptosis., 2008, 13, 11-32.
- A. Chiarugi, FASEB J., 2005, 19, 1783-1788.
- I.T. Abdel-Raheem, A.A. Abdel-Ghany, G.A. Mohamed, Biol. Pharm. Bull., 2009, 32, 61-67.



