Review Article - Der Pharma Chemica ( 2022) Volume 14, Issue 12
Review on Therapeutic Uses , Side Effects , Pharmacokinetics and Drug Interaction of Some Antibiotics
Biplab De1 and Durba Bannerjee2*2Department of Pharmaceutical Chemistry, Guru Nanak Institute of Pharmaceutical Science and Technology, E157/F, Nilgumj Rd, Sahid Colony, Panihati, Kol, India
Durba Bannerjee, Department of Pharmaceutical Chemistry, Guru Nanak Institute of Pharmaceutical Science and Technology, E157/F, Nilgumj Rd, Sahid Colony, Panihati, Kol, India, Email: banerjeedurba34@gmail.com
Received: 25-Nov-2022, Manuscript No. dpc-22-81326,; Editor assigned: 28-Nov-2022, Pre QC No. dpc-22-81326,; Reviewed: 12-Dec-2022, QC No. dpc-22-81326,; Revised: 14-Dec-2022, Manuscript No. dpc-22-81326,; Published: 21-Dec-2022, DOI: 10.4172/0975-413X.14.12.1-10
Abstract
An antibiotic is a type of antimicrobial substance active against bacteria. They may either kill or inhibit the growth of bacteria. A limited number of antibiotics also possess antiprotozoal activity. Antibiotics are not effective against viruses. History, classification, mechanism of action, drug interaction, clinical uses, doses, side effects, pharmacokinetics etc. of antibiotics are elaborately discussed.
Keywords
Antibiotics; Pharmacokinetics; Drug interaction
INTRODUCTION
We usually associate the beginning of the modern “antibiotic era” with the names of Paul Ehrlich and Alexander Fleming. Ehrlich's idea of a “magic bullet” that selectively targets only disease-causing microbes and not the host was based on an observation that aniline and other synthetic dyes, which first became available at that time, could stain specific microbes but not others. Ehrlich argued that chemical compounds could be synthesized that would “be able to exert their full action exclusively on the parasite harbored within the organism.” This idea led him to begin a large-scale and systematic screening program (as we would call it today) in 1904 to find a drug against syphilis, a disease that was endemic and almost incurable at that time. This sexually transmitted disease, caused by the spirochete Treponema pallidium, was usually treated with inorganic mercury salts but the treatment had severe side effects and poor efficacy. In his laboratory, together with chemist Alfred Bertheim and bacteriologist Sahachiro Hata, they synthesized hundreds of organ arsenic derivatives of a highly toxic drug Atoxyl and tested them in syphilis-infected rabbits. In 1909 they came across the sixth compound in the 600th series tested, thus numbered 606, which cured syphilis-infected rabbits and showed significant promise for the treatment of patients with this venereal disease in limited trials on humans . Despite the tedious injection procedure and side effects, the drug, marketed by Hoechst under the name Salvarsan, was a great success and, together with a more soluble and less toxic Neosalvarsan, enjoyed the status of the most frequently prescribed drug until its replacement by penicillin in the 1940s. Amazingly, the mode of action of this 100-year-old drug is still unknown, and the controversy about its chemical structure has been solved recently.
Probably many of us are familiar with the somewhat serendipitous event on the September 3, 1928 that led to the penicillin discovery by Fleming. Although the antibacterial properties of mold had been known from ancient times, and researchers before him had come upon the similar observations regarding the antimicrobial activity of Penicillium from time to time, it was his formidable persistency and his belief in the idea that made the difference. For 12 years after his initial observation, A. Fleming was trying to get chemists interested in resolving persisting problems with purification and stability of the active substance and supplied the Penicillium strain to anyone requesting it. He finally abandoned the idea in 1940, but, fortunately, in the same year an Oxford team led by Howard Florey and Ernest Chain published a paper describing the purification of penicillin quantities sufficient for clinical testing. Their protocol eventually led to penicillin mass production and distribution in 1945. Fleming's screening method using inhibition zones in lawns of pathogenic bacteria on the surface of agar-medium plates required much less resources than any testing in animal disease models and thus became widely used in mass screenings for antibiotic-producing microorganisms by many researchers in academia and industry. Fleming was also among the first who cautioned about the potential resistance to penicillin if used too little or for a too short period during treatment [1].
An antibiotic is a type of antimicrobial substance active against bacteria. It is the most important type of antibacterial agent for fighting bacterial infections, and antibiotic medications are widely used in the treatment and prevention of such infections. They may either kill or inhibit the growth of bacteria. A limited number of antibiotics also possess antiprotozoal activity. Antibiotics are not effective against viruses such as the common cold or influenza; drugs which inhibit viruses are termed antiviral drugs or antivirals rather than antibiotics. Chemically antibiotics can be classified as follows [2] (Figures 1-7).

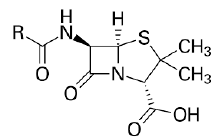
Figure 1: β- lactam antibiotics.

Figure 2: Macrolide antibiotics.

Figure 3: Tetracyclines.

Figure 4: Nitrobenzene derivative.

Figure 5: Aminoglycosides.

Figure 6: Quinolones and fluoroquinolones.

Figure 7: Sulphonamides.
CLASSIFICATION OF KNOWN ANTIBIOTICS IN GENERAL
Some antibiotics are discussed with their chemical structure and common probable mechanism of action [3-9] in the table 1.
| Sl. No. | Classified Group | Name Of Antibiotics | General Chemical Structure | Common Probable Mechanism Of Action |
|---|---|---|---|---|
| 1. | Beta Lactam Antibiotics | Penicillin: Penicillin G, Penicillin V, Oxacillin, Cloxacillin, Carbenicillin Piperacillin, Ampicillin, Amoxicillin. |
|
Beta lactam antibiotic kills susceptible bacteria by specifically inhibiting the transpeptidase that catalyzes the final step in cell wall biosynthesis, the cross-linking of peptidoglycan. |
| Cephalosporin: Cefepime, ceftriaxone, cefuroxime, cefazolin, cefadroxil, cefuroxime, Cefdinir. | 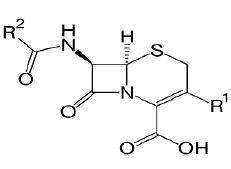 |
|||
| Carbapenams: Imipenem, Biapenam. | 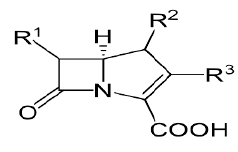 |
|||
| Monobactam: Aztreonam | 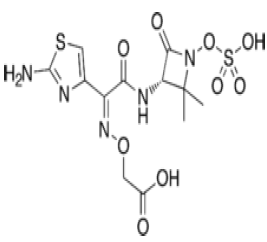 |
|||
| 2. | Aminoglycoside Antibiotics | Gentamicin, Amikacin, Tobramycin, Neomycin, and Streptomycin. | 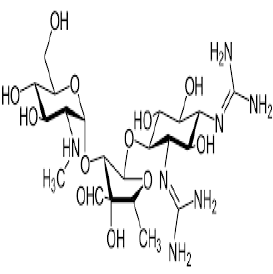 |
The aminoglycosides primarily act by binding to the aminoacyl site of 16S ribosomal RNA within the 30S ribosomal subunit, leading to misreading of the genetic code and inhibition of translocation in bacteria. |
| 3. | Macrolide Antibiotics | erythromycin, clarithromycin, azithromycin, fidaxomicin and telithromycin | 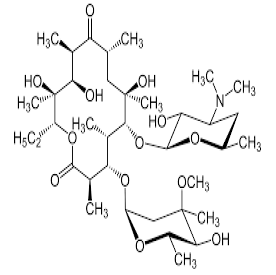 |
Macrolies inhibit bacterial protein synthesis. The mechanism of action of macrolides revolves around their ability to bind the bacterial 50S ribosomal subunit causing the cessation of bacterial protein synthesis. |
| 4. | Tetracycline Antibiotics | Short acting: chlortetracycline, oxytetracycline. | 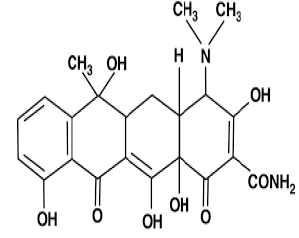 |
Tetracycline antibiotics are protein synthesis inhibitors . They inhibit the initiation of translation in variety of ways by binding to the 30S ribosomal subunit, which is made up of 16S rRNA and 21 proteins. They inhibit the binding of aminoacyl-tRNA to the mRNA translation complex. |
| Intermediate acting: demeclocycline, lymecycline | ||||
| Long acting: minocycline, doxycycline. | ||||
| 5. | Fluoroquinolone Antibiotics | Ciprofloxacin, ofloxacin. | 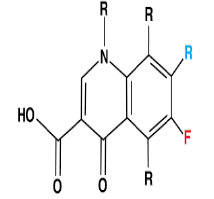 |
The fluoroquinolones are bactericidal and target topoisomerase, or DNA gyrase, a substance that functions to preserve the state of supercoiling in replicating and non-replicating bacterial chromosomes. |
| 6. | Lipopetide Antibiotics |
|
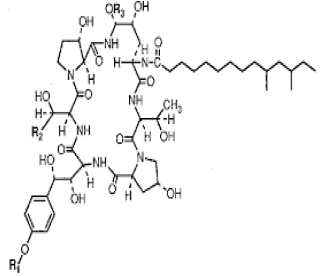 |
Lipopeptides exert their effect by binding and disrupting the cell membrane integrity of the target bacteria and initiating a series of events that eventually leads to cell death |
| 7. | Sulfonamide Antibiotics | Sulfadiazine, sulfamethizole, sulfamethoxazole, sulfasalazine, sulfisoxazole. | 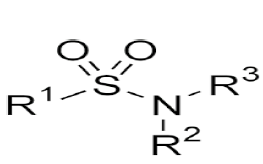 |
Competitively inhibit the incorporation of PABA into folic acid, thereby preventing the synthesis of folic acid. Trimethoprim binds reversibly to and inhibits dihyrofolate reductase, an enzyme that reduces dihydrofolic acid to tetrahydrofolic acid,decreasing folic acid synthesis in bacteria. |
DOSE, THERAPEUTIC USES AND SIDE EFFECTS OF FEW ANTIBIOTICS
Some antibiotics [3-10] with their dose, therapeutic uses and side effects are discussed in the table 2.
| Sl. No. | Name | Dose | Therapeutic Uses | Side Effects |
|---|---|---|---|---|
| 1 | Amoxicillin | 500mg/dose | To treat certain infections caused by bacteria, such as pneumonia, bronchitis (infection of the airway tubes), H. pylori infection and duodenal ulcers. | Back, leg or stomach pain, abdominal or stomach cramps, bloating. |
| 2 | Amoxicillin + Clavonic acid | 2.4 gm + 600 mg /day | To treat certain infections caused by bacteria, including infections of the ears, lungs, sinus, skin, and urinary tract. | Pale stools, dark urine, loss of appetite, nausea, vomiting. |
| 3 | Ampicillin | 2-3 gm/day | Meningitis (infection of the membranes that surround the brain and spinal cord), and infections of the throat, sinuses, lungs, reproductive organs, urinary tract, and gastrointestinal tract. | Difficulty in breathing or swallowing. |
| 4 | Penicillin | 24 million units /day | To treat certain infections caused by bacteria such as pneumonia and other respiratory tract infections, scarlet fever, and ear, skin, gum, mouth, and throat infections . | Mild nausea or diarrhea, headache, or vaginal itching. |
| 5 | Cloxacillin | 4 gm /day | To treat infections of the skin, bone, heart valve, blood, and lung. | Upset stomach, nausea, vomiting, diarrhea, gas, and mouth sores. |
| 6 | Cephalaxin | 2gm /day | To treat certain infections caused by bacteria such as pneumonia and other respiratory tract infections; and infections of the bone, skin, ears, , genital, and urinary tract. |
|
| 7 | Cefixime | 400 mg /day | Otitis media, strep throat, pneumonia, urinary tract infections, gonorrhea, and Lyme disease. |
|
| 8 | Azithromycin | 500 mg /day | Bronchitis, pneumonia, sexually transmitted diseases (STD), and infections of the ears, lungs, sinuses, skin, throat, and reproductive organs. | Stomach upset, diarrhea/loose stools, nausea, vomiting, or abdominal pain. |
| 9 | Erythromycin | 4 gm /day | Respiratory tract infections, skin infections, diphtheria, intestinal amebiasis, acute pelvic inflammatory disease, Legionnaire's disease, pertussis, and syphilis. | Nausea, vomiting, diarrhea, stomach pain/cramping, and loss of appetite. |
| 10 | Clarithromycin | 1 gm /day | Pneumonia (a lung infection), bronchitis (infection of the tubes leading to the lungs), and infections of the ears, sinuses, skin, and throat. | Diarrhea, nausea, vomiting, headache, and changes in taste. |
| 11 | Doxycycline | 200 mg /day | To treat pimples and abscesses (usually on the face) that is caused by rosacea, also known as acne rosacea or adult acne. | Loss of appetite, nausea and vomiting, diarrhea, rash, sensitivity to the sun. |
| 12 | Tetracycline | 3 gm /day | Pneumonia and other respiratory tract infections; certain infections of skin, eye, lymphatic, intestinal, genital and urinary systems; and certain other infections that are spread by ticks, lice, mites, and infected animals. | Nausea, vomiting, diarrhea, loss of appetite, mouth sores, black hairy tongue, sore throat. |
| 13 | Ciprofloxacin | 1 gm /day | Pneumonia skin, bone infections, gonorrhea. | Blistering, peeling, or loosening of the skin · bluish-colored lips. |
| 14 | Ofloxacin | 400 mg /day | Bronchitis, pneumonia, chlamydia, gonorrhea, skin infections, urinary tract infections, and infections of the prostate . |
|
| 15 | Metronidazole | 600 mg /day | To treat infections of the reproductive system, gastrointestinal (GI) tract, skin, heart, bone, joint, lung, blood, nervous system, and other areas of the body. | Feeling or being sick, stomach pain, hot flushes, difficulty breathing, a pounding heartbeat (palpitations) and headaches. |
| 16 | Clotrimazole | 3.8 gm /day | To treat skin infections such as athlete's foot, jock itch, ringworm, and other fungal skin infections (candidiasis). | Burning, stinging, swelling, irritation, redness, pimple-like bumps, tenderness. |
| 17 | Clindamycin | 1.2-1.8 gm /day | Infections of the lungs, skin, blood, female reproductive organs, and internal organs. | Nausea, vomiting, stomach pain, mild skin rash or vaginal itching or discharge. |
PHARMACOKINETICS AND PROBABLE DRUG INTERACTIONS OF ANTIBIOTICS [3- 10]
β-lactam antibiotics
The oral β-lactams are an extremely well-absorbed class of antibiotics. They share a common uptake mechanism with small peptides for entry into the enterocyte and exit through the basolateral membrane. Transport proteins involved in the absorption of these antibiotics have been identified. Three strategies were employed to identify proteins important for the proton dependent uptake of β-lactams. First, a 127-kDa β-lactam transporter in rabbit intestinal brush border membranes was identified by photoaffinity labeling, protein purification and reconstitution. Second, an immunological approach resulted in the identification of HPT-1, a 120-kDa protein in apical membranes of human intestinal Caco-2 cells. The hpt-1 cDNA confers the ability to transport both cephalexin and bestatin in a proton-dependent fashion. Third, functional cloning of poly(A)+ RNA of rabbit intestine identified a PepT1 protein that cotransports protons with small peptides, β-lactams, and angiotensin-converting enzyme inhibitors. The kidney also expresses a transporter, PepT2, with high homology to PepT1 [11]. The serum half-life (t1/2) of most beta-lactams is 1-2 hours. Ceftazidime and temocillin are more long-lasting with a t1/2 of 4-6 hours and ceftriaxone of 8-10 h. These values relate to patients with normal renal function. The elimination is only moderately prolonged for beta-lactams and of little consequences for the dosage regimens, partly because of the high tolerance of this group of antibiotics. Penicillins and cephalosporins are eliminated by glomerular filtration and varying degrees of active transport across the epithelial cells of the renal tubuli and hepatobiliary system. Active transport mechanisms also explain the low concentrations of beta-lactam antibiotics in cerebrospinal fluid. Otherwise, penetration of these agents to unspecialized tissues is good. Substances with a low serum protein binding, such as ampicillin and amoxicillin, reach concentrations in peripheral human lymph which as assessed by the ratios of areas under the concentration curves are 50-80% of the serum levels. The serum protein binding appears to act mainly by inhibiting the rate with which the antibiotic passes into extravascular foci. For instance, temocillin with a serum albumin binding around 85% establishes levels in peripheral lymph which are 50-60% of those in serum. This may be explained by the fact that a higher protein binding is accompanied by more lipid solubility, which are two factors acting in opposite directions as concerns passage by diffusion across body barriers.10Penicillins+Aminoglycosides: There is an in vitro interction between aminoglycoside antibiotics and penicillins leading to a significant loss of aminoglycoside antibacterial activity if these antibiotics are mixed in the same bottle [12].
Aminoglycoside antibiotics
The aminoglycosides are poorly absorbed from the gastrointestinal tract and therefore are given parenteral, via intramuscular or intravenous injection, or topically, via application to the skin. In the neonatal period, the absorption rate after intramuscular injection appears to be faster than reported in adults and the volume of distribution is significantly larger. The major pharmacokinetic difference between neonates, infants and adults is the slower elimination: for instance, half-lives which average around 2 hours in adults with normal renal function can reach and sometimes exceed 5 to 6 hours during the very first days of life. The volume of distribution per kilogram of body weight is relatively smaller in obese subjects and in dehydrated patients than in normal subjects and can lead to overdosing when dosages are calculated on a body weight basis. Anemia, fever, hypoxaemia and major burns are other pathological states which significantly influence pharmacokinetics of aminoglycosides. Aminoglycosides are neither protein bound nor biotransformed. The major route of elimination is glomerular filtration, and aminoglycosides undergo some tubular reabsorption. Due to the almost exclusive excretion from the body by glomerular filtration, the elimination rate of aminoglycoside antibiotics is greatly affected by impairment of renal function. Due to the almost exclusive excretion from the body by glomerular filtration, the elimination rate of aminoglycoside antibiotics is greatly affected by impairment of renal function.Recent studies have shown that a combination of penicillin plus gentamicin produces enhanced killing against virtually all strains of enterococci in vitro.Administration of an aminoglycoside followed by furosemide may increase the risk of ototoxicity. The aminoglycoside interacts with the cell membranes in the inner ear, increasing their permeability.Aminoglycoside antibiotics can damage the vestibular and auditory sensory epithelia, and the loop diuretics can enhance the ototoxic effect of aminoglycosides. Thus aminoglycocide are not recommended when loop diuretics are being administerd .It should not be taken by mouth or intravenously if anyone is already taking theracrys (BCG live intravesical) or Vistide (cidofovir) or Zanosar (streptozocin) [13].
Macrolide antibiotics
Macrolide antibiotics are basic compounds, poorly soluble in water, which are mostly absorbed in the alkaline intestinal environment. They are acid unstable, but the newer semisynthetic derivatives (i.e. roxithromycin and azithromycin) are characterised by increased stability under acidic conditions. Macrolides are highly liposoluble and consequently penetrate well into tissue, especially bronchial secretions, prostatic tissue, middle ear exudates and bone tissues, as evidenced by tissue/serum concentration ratios greater than 1. They do not penetrate well into the CSF. Macrolides undergo extensive biotransformation in the liver. With a few exceptions (e.g. miocamycin), the metabolites of these drugs are characterised by little or no antimicrobial activity. Plasma protein binding is variable from one compound to another. At therapeutic concentrations, protein-bound erythromycin accounts for 80 to 90% of the total drug present in the blood, and the fraction is 95% for roxithromycin. The lowest values of protein-bound fraction are observed for midecamycin and josamycin (about 15%), and intermediate values are reported for spiramycin and miocamycin. However, the clinical relevance of this parameter is not clearly established. Plasma half-life (t1/2) values vary for the macrolides described: erythromycin, oleandomycin, josamycin and miocamycin have a t1/2 ranging from 1 to 2 hours; spiramycin, erythromycin stearate, the mercaptosuccinate salt of propionyl erythromycin and rosaramicin have an intermediate t1/2 (about 7, 6.5, 5 and 4.5 hours, respectively); the newer semisynthetic compounds roxithromycin and azithromycin are characterised by high t1/2 values (i.e. 11 and 41 hours, respectively). Under normal conditions, the major route of elimination is the liver. Renal elimination also takes place but it contributes to total clearance only to a small degree, as evidenced by low renal clearance values. The degree of modification of macrolide pharmacokinetics by renal insufficiency or hepatic disease is usually not considered clinically relevant, and no recommendation for dose modification is necessary in these patients [14]. The macrolides are inactivated in the liver, and the major route of elimination is in the bile. Drug-drug interactions of macrolide antibiotics [15] are represented in the table 3.
| Azithromycin shows with | Clarithromycin shows with | Dirithromycin shows with | Erithromycin shows with | Roxithromycin shows with |
|---|---|---|---|---|
| Antacids | Astemizole | Astemizole | Astemizole | Cyclosporin |
| Cyclosporin | Carbamazepine | Cyclosporin | Benzodiazepines | Digoxin |
| Digitoxin | Cimitidine | Digoxin | Buspirone | Theophylline |
| Lovastatin | Cyclosporin | Food | Carbamazepine | |
| Rifabutin | Deigoxin | Oral contraceptives | Cisapride | |
| Theophylline | Fluoxitine | Theophylline | Clozaine | |
| Warfarin | Indinavir | Cyclosporin | ||
| Loratadine | Digoxin | |||
| Midazolam | Felodipine | |||
| Pimozide | Fexofenadine | |||
| Rifabutin | HMG-CoA inhibitor | |||
| Ritonavir | Methylprednisolone | |||
| Saquinavir | Tacrolimus | |||
| Terfenadine | Terfenadine | |||
| Theophylline | Theophylline | |||
| Verapamil | Vinblastin | |||
| Warfarin | Warfarin | |||
| Zidovudine |
Tetacycline antibiotics
Bioavailability is less than 40% when administered via intramuscular injection, 100% intravenously and 60-80% orally (fasting adults). Food and/or milk reduce GI absorption of oral preparations of tetracycline by 50% or more. Absorption is variable ranging from 0% to almost 90%; however, for most agents it is in the range 25-60%. Serum concentrations rise slowly after oral administration with absorption occurring in the stomach, duodenum and small intestine. Cmax (mg/L) depends on dose, but is generally in the range 1–5 mg/L. tmax is in the range 2-4 h except for demeclocycline whose Cmax is delayed until 4-6 h [16]. All tetracyclines form insoluble complexes with calcium, magnesium, iron and aluminium, which markedly reduce absorption [17]. The effect of disease on the absorption of these drugs is unknown. Protein, fat and carbohydrate meals reduce the absorption of tetracycline by about 50%. The volume of distribution (V) for these agents is in the order of 1.3–1.7 L/kg or a total volume of distribution of 100-130 L. These data imply some concentration in tissues; however, most data on tissue penetration are of poor quality, making firm conclusions about their relative distribution difficult. Protein binding is variable. None of these agents undergoes metabolism with the exception of tetracycline, 5% of which is excreted as the metabolite Δ-epitetracycline. Unchanged drugs are excreted by renal and bilary routes. Renal elimination (CLR) is related to glomerular filtration for most agents, the exception being chlortetracycline.The amount of drug excreted in the urine is <50%; rolitetracycline is said to have high renal elimination. Greater than 40% appears in the faeces after biliary elimination and most drugs have some enterohepatic circulation. Antacids containing magnesium, aluminum, calcium, or sodium bicarbonate, calcium supplements, zinc products, iron products, and laxatives containing magnesium interfere with tetracycline, making it less effective. Tetracyclines are contraindicated in pregnancy because of the risk of hepatotoxicity in the mother, the potential for permanent discoloration of teeth in the fetus (yellow or brown in appearance), as well as impairment of fetal long bone growth. tetracyclines are bacteriostatic in action and penicillins act on the cell wall of actively growing bacteria they should not be used together, as the former will stop the latter working.
Quinolone and fluoroquinolone antibiotics
Ofloxacin, levofloxacin, pefloxacin, fleroxacin, temafloxacin and lomefloxacin are almost completely absorbed, while ciprofloxacin and enoxacin have a lower bioavailability. The absorption of N′4-methylated compounds, such as ofloxacin, levofloxacin, pefloxacin and fleroxacin, tends to be greater than that of non-N′4-methylated compounds. At the acidic pH of the stomach, all fluoroquinolones are protonated and exist as the cationic species. Although the solubility of most fluoroquinolones is still not high at the slightly alkaline pH of the jejunum, they are well absorbed in the duodenum and jejunum, possibly because the percentage of the more lipophilic neutral species of the agent increases. The critical role of dissolved drug suggests that the absorption kinetics of fluoroquinolones may be affected to a great degree by their solubility and dissolution rates. This is supported by evidence that the Tmax of temafloxacin formulations increases with dose. After oral administration, both the maximum serum concentration (Cmax) and time to reach this concentration (Tmax) are affected primarily by the absorption constant. ost fluoroquinolones are excreted primarily by the kidney but the gastrointestinal route also plays a significant role in the overall elimination of these agents. Hepatic elimination plays a role only for certain agents such as pefloxacin. The physicochemical properties of fluoroquinolones are greatly influenced by their two functional groups, the carboxyl group and basic group, which affect pH-dependent lipophilicity. Many important properties of distribution may be explained by interactions between the pH of these groups and the local pH in the body. These physico-chemical properties may also partly explain the elimination properties of fluoroquinolones. They all undergo tubular secretion as either acids or bases and some are also significantly reabsorbed. Hepatic handling and resultant metabolite excretion is also influenced by N′4-methylation. The quinolone antibacterials are prone to many interactions with other drugs. Quinolone absorption is markedly reduced with antacids containing aluminium, magnesium and/or calcium and therapeutic failure may result. Other metallic ion-containing drugs, such as sucralfate, iron salts, and zinc salts, can also reduce absorption. Some of the newer quinolones inhibit the cytochrome P450 system, e.g. enoxacin, pefloxacin and ciprofloxacin. The toxicity of drugs that are metabolised by the cytochrome P450 system is enhanced by concomitant use of some quinolones. Ciprofloxacin, enoxacin and pefloxacin can increase theophylline concentrations to toxic values. The pharmacokinetics of warfarin and cyclosporin are unaffected. Ofloxacin, fleroxacin and temafloxacin have a low inhibitory effect on the cytochrome P450 system and a low interaction potential may result. The affinity of quinolones for the gamma-aminobutyric acid (GABA) receptor may induce CNS adverse effects; these effects are enhanced by some nonsteroidal anti-inflammatory drugs (NSAIDs) [18].
Sulfonamide antibiotics
Most sulfonamides are readily absorbed orally and, when applied to burns, topically. Sulfonamides are distributed throughout the body. They are metabolized mainly by the liver and excreted by the kidneys. Sulfonamides compete for bilirubin-binding sites on albumin. Sulfonamides may displace from albumin-binding sites drugs such as warfarin, increasing the effective activity of the displaced drug. Anticoagulant dosage should be reduced during sulfonamide therapy. Sulfonamides also displace methotrexate from its bound protein, increasing methotrexate toxicity. An increased hypoglycemic effect of chlorpropamide and tolbutamide may occur during sulfonamide therapy, possibly because of the same mechanism or structural similarities. Sulfonamides may compete for binding sites with some anesthetic agents such as thiopental, and reduced barbiturate doses might be necessary. Sulfonamides may potentiate the action of some thiazide diuretics, phenytoin, and uricosuric agents. Conversely, sulfonamides themselves can be displaced from binding sites by indomethacin, phenylbutazone, salicylates, probenecid, and sulfinpyrazone, resulting in increased sulfonamide activity. Cyclosporine levels may be reduced by sulfonamides. Oral contraceptive failure during sulfonamide therapy has been noted rarely [19,20].
Lipopeptide antibiotics
Lipopeptides are not absorbed from the gastrointestinal tract and needs to be administered parenterally. The distribution of lipopeptides are limited (volume of distribution of 0.1 L/kg in healthy volunteers) due to its negative charge at physiological pH and its high binding to plasma proteins (about 90%). Their elimination is mainly renal, with about 50% of the dose excreted unchanged in the urine. Ibuprofen increases the risk of renal impairment when given with lipopeptides such as Daptomycin. Daptomycin has moderate interactions with the following drugs: atorvastatin, fluvastatin, lovastatin, pitavastatin, pravastatin, rosuvastatin, simvastatin [21].
CONCLUSION
Antibiotics such as bactericidal and bacteriostatic drugs are generally used to treat different type of bacterial infections. Antibiotics should not be taken without consultation with registered medical practitioner considering their chemical structure, mechanism of action, side effects and dose fixation. Self-medication in use of antibiotics may be dangerous. Antibiotics ultimately can be characterized as lifesaving drugs.
REFERENCES
- Hutchings MI, Trueman AW, Wilkison B. Curr Opin Microbiol. 2019, 51(10): p. 72-80.
- Morier D. Encyclopaedia Britannica. 2022.
- Yocum RR, Rasmussen JR, Strominger JL. J Biochem. 1980, 255(9): p. 3977-3986.
- Bush K, Bradford PA. Cold Spring Harb Perspect Med. 2016, 6(8): p. 25-47.
- Kumana CR, Yuen KY. Drugs. 1994, 47: p. 902- 904.
- Patel PH, Hashmi MF. Macrolides. In: StatPearls. Treasure Island (FL): 2022.
- Fàbrega A, Madurga S, Giralt E, et al., Microb Biotechnol. 2009, 2(1): p. 40-61.
- Memon N. How do lipopeptides work? RXList. 2022.
- Bambeke FV, Paul MT. Sulfonamides. Infectious Diseases (Fourth Edition), Science Direct, 2017.
- Usual dosages of commonly prescribed antibiotics, MSD manual professional version.
- Dantzig AH. Adv Drug Deliv Rev. 1997, 23(1): p. 63-76.
- Bergan T. Scand J Infect Dis Suppl. 1984, 42: p. 83-98.
- Pechere JC, Dugal R. Clin Pharmacokinet. 1979, 4: p. 170-199.
- Periti P, Mazzei T, Mini E, et al., Clin Pharmacokinet. 1989, 16(4): p. 193-214
- Pal M, Graci D, Amsden G. Ann Pharmacother. 2000, 34(4): p. 495-513
- Agwuh KN, MacGowan A. J Antimicrob Chemother. 2006, 58(2): p. 256-265.
- Shutter MC, Akhondi H. Tetracycline. Treasure Island (FL). 2022.
- Turnidge J. Drugs. 1999, 58(2): p. 29-36.
- Brouwers JR. Drug Saf. 1992, 7(4): p. 268-81.
- Vree TB, Hekster YA. Karger. 1985.
- Dvorchik B, Damphousse D. J Clin Pharmacol. 2004, 44(6): p. 612-20.
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref



