Research Article - Der Pharma Chemica ( 2023) Volume 15, Issue 2
Study of The Chemical Reactivity of a Series of Halogen-Substituted Imidazole-Thiosemicarbazides Using Density Functional Theory
Mamadou Guy-Richard KONE1,2,3*, Georges Stephane DEMBELE1,2, Bafetigue OUATTARA4, Adama NIARE4, Panaghiotis KARAMANIS3 and Nahosse ZIAO1,22Groupe Ivoirien de Recherches en Modelisation des Maladies (GIR2M), Universite NANGUI ABROGOUA, Abidjan, Cote D'Ivoire
3E2S UPPA, CNRS, IPREM, Université de Pau et des Pays de l’Adour, 64053 Pau, France
4Laboratoire de Physique Fondamentale et Appliquee, UFR SFA, Universite NANGUI ABROGOUA, Abidjan, Cote D'Ivoire
Mamadou Guy-Richard KONE, Laboratoire de Thermodynamique et de Physico-Chimie du Milieu, Universite NANGUI ABROGOUA, Abidjan, Cote D'Ivoire, Email: guyrichardkone@gmail.com
Received: 30-Jan-2023, Manuscript No. dpc-23-88268; Editor assigned: 01-Feb-2023, Pre QC No. dpc-23-88268; Reviewed: 15-Feb-2023, QC No. dpc-23-88268; Revised: 17-Mar-2023, Manuscript No. dpc-23-88268; Published: 24-Feb-2023, DOI: 10.4172/0975-413X.15.2.1-10
Abstract
This theoretical reactivity study was conducted on six molecules of a series of halogen-substituted Imidazole-Thiosemicarbazides (ITS) using density functional theory, at the B3LYP/6-31+G (d, p) level. Analysis of the thermodynamic formation quantities confirmed the formation and existence of the series of molecules studied. The study of the boundary molecular orbitals, including the energy gap (ΔE), electronegativity (?), chemical hardness (η) and electrophilicity index (ω) provided a better overview of the molecular properties. Thus, the compounds ITS 1 and ITS 4 which have the lowest energy gaps between the boundary orbitals are the most reactive and the least stable. Furthermore, ITS 1 is the softest of the compounds studied. The analysis of the local descriptors and the isodensity map allowed us to identify the N14 nitrogen atom as the preferred electrophilic attack site and the C18 carbon atom as the preferred nucleophilic attack site. These electrophilic and nucleophilic attack sites (N14 and C18) are identical for all compounds according to the dual descriptors. Furthermore, halogen substitution on imidazole-thiosemicarbazides does not change the centres of reactivity. The dendrogram of the Hierarchical Ascending Classification Analysis allowed us to group all the six studied compounds into three categories. The most active one is ITS 3, ITS 2 and ITS 5 the moderately active compounds and ITS 1, ITS 4 and ITS 6, the least active ones. The surface profiler analysis showed us an almost smooth plane connecting the three (3) descriptors that are ΔE, ω and η. This result shows the linearity between these descriptors of reactivity.
Keywords
Chemical reactivity; Global descriptors; Local descriptors; Dual descriptors
Introduction
Parasites from plants or animals origin, feed themselves at the expense of a host without which they are unable to survive. Parasitism is a universally widespread phenomenon, which affects practically all living creatures. For example, we can quote parasites that attack humans such as lice, fleas and intestinal worms. It is in this context that Agata et al. [1] have synthesized and tested imidazole-thiosemicarbazides with halogens as substitution to fight against infection caused by Toxoplasma gondii. It should be noted that Toxoplasma gondii is a species of intracellular parasites belonging to the phylum Apicomplexa, and it is the pathogen agent of toxoplasmosis. The Apicomplexa phylum includes many other pathogens of medical or veterinary importance; among them we can quote Plasmodium falciparum which is responsible for malaria in humans. In addition, this parasite affects around 30% of the world population [2]. It causes serious illnesses in people living with HIV/AIDS or in pregnant women creating birth defects [3,4]. Imidazole-thiosemicarbazides are practical precursors that have been widely used in heterocyclic synthesis. One of the most complex branches of organic chemistry is the chemistry of heterocyclic compounds. It is also interesting for its theoretical implications, because of the diversity of its synthesis methods, and because of the physiological and industrial significance of heterocyclic compounds. Studies on heterocyclic compounds have a longtime been an interesting area in medicinal chemistry. For better understanding of the ways how to control these harmeful and mortal parasites, medicinal chemistry or therapeutic chemistry, which is a scientific discipline at the interface of chemistry and pharmacy, including the design of drugs and their development, is on the lookout for new molecular entities with biological or therapeutic activity. Nowadays, computational chemistry gives a lot of information on the electronic structures of molecules and contributes largely to the development of traditionally experimental chemistry [5-7]. In this work, a series of six molecules of Imidazole-Thiosemicarbazides Substituted (ITS) by halogens like Chlorine and Fluorine in Ortho, Meta and Para position (ITS 1, ITS 2, ITS 3, ITS 4, ITS 5 and ITS 6) have been used (Figure 1). The aim of this work is to theoretically determine, on the one hand the reactivity of imidazole-thiosemicarbazides substituted by halogens and on the other hand to identify the sites of nucleophilic / electrophilic attacks by different methods of quantum chemistry.

Figure 1. Structures and nomenclature of studied Imidazole-Thiosemicarbazides and their codes
MATERIAL AND METHODS
Calculation theory level
The theoretical study of chemical reactivity was carried out based on three theoretical approaches. The first concerns the analysis of molecular isodensity maps. The second one relates to border molecular orbitals. And finally the last approach deals with local indices of reactivity as well as dual descriptors. The geometries of the molecules have been optimized at the DFT calculation level with the functional B3LYP [8-10] in the base 6-31 + G (d, p) using the Gaussian software 09 [11]. This hybrid functional gives better energies and is in agreement with high calculations level of ab initio methods [12, 13]. As for the base split-valence and double-dzéta (6-31G (d, p)), it is sufficiently extended and the fact of taking into account the functions of polarization is important for the explanation of the free doublets of the heteroatoms. The geometries are kept constant for cationic and anionic systems. The global reactivity indices were obtained from the conceptual DFT model [14-16]. The Hierarchical Ascending Classification (HAC) analysis was carried out using XLSTAT software [17]. As for the local chemical reactivity indices, they were determined using the electronic populations calculated with the Hirshfeld Population Analysis (HPA) [18].
Thermodynamic parameters of formation
The thermodynamic quantities of the studied molecules were carried out from optimization and the frequency calculation at the level B3LYP / 6-31 + G (d, p). The magnitudes such as the entropy, the enthalpy and the free enthalpy of ITS’ formation were determined using the following formulas proposed by Otchersky et al. [11].
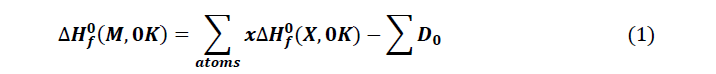
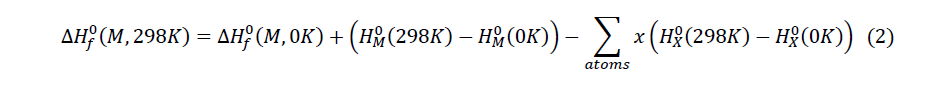
With
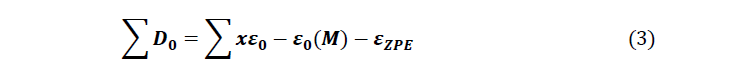

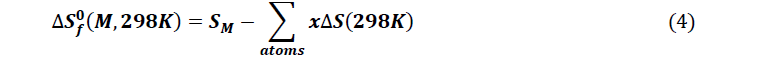
x : Number of atoms of X in the Molecule
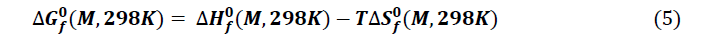
Hirshfeld Population Analysis
Hirshfeld charges analysis has been used extensively, particularly for the calculation of Fukui coefficients. Before obtaining Hirshfeld charges of a molecule that one must first be decomposed into atomic fragments. A general and natural choice consist to share the charge density at each point between the different atoms in proportion to their densities of free atoms at the corresponding distances from the nuclei [20,8]. This method allows obtaining localized and bound electronic distributions, which is approximately like the molecular electronic density. The integration of the densities surrounding each atom defines its net atomic charge.
Reactivity descriptors
Global descriptors
To predict chemical reactivity, some theoretical descriptors related to conceptual DFT have been determined. In particular, the energy of the lowest unoccupied Molecular Orbital (LUMO), the energy of the Highest Occupied Molecular Orbital (HOMO), electronegativity (χ), global softness (σ) and global electrophilicity index (ω). These descriptors are all determined from the optimized structure of the molecules. It should be noted that, the descriptors related to the molecular orbital boundaries have been calculated in a very simple way within the framework of Koopmans’ approximation [21].
LUMO energy characterizes the sensitivity of the molecule to nucleophilic attack, and HOMO energy characterizes the susceptibility of a molecule to electrophilic attack. The electronegativity (χ) is the parameter that reflects the ability of a molecule not to let its electrons escape. Global softness (σ) expresses the resistance of a system to changes in its number of electrons. The global electrophilicity index characterizes the electrophilic power of the molecule. These different parameters are calculated from equations (6):
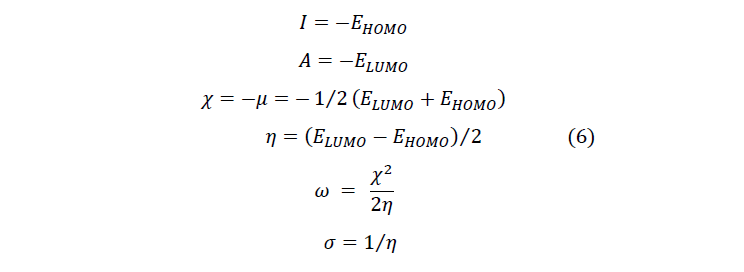
Local and dual descriptors
The Fukui indices of a molecule inform about the local reactivity in a molecule. The atom with the highest value of Fukui index is more reactive than the other atoms belonging to the molecule [22]. These indices represent the qualitative description of the reactivity of the atoms in the molecule. The Fukui function successfully predicts relative reactivity for most chemical systems. Fukui indices for the selectivity of electrophilic and nucleophilic atoms in Imidazole-Thiosemicarbazides compounds have been determined. Ayers and Parr [23] explained that molecules tend to react where the Fukui function is greatest when they are attacked by soft reagents and similarly where the Fukui function is smallest it is where it will be attacked by hard reagents. Using the Hirshfeld atomic charges of the optimized compounds at the ground state, the Fukui functions (fk+,fk−), the local softness (sk+,sk−) and the local electrophilic indices (ωk+,ωk−) [24] were determined. The Fukui functions are calculated using equations (7) and (8):


Local softness and electrophilicity indices are calculated using (9-12)


RESULTS AND DISCUSSION
The study of the reactivity of organic molecules is essential in the pharmaceutical, cosmetic and food-processing fields. In these fields, the prediction of the evolution of product quality under the influence of external factors is important. These factors include temperature, humidity, light, oxygen and pH. They permit to define the storage and transportation conditions and the expiry date of these organic compounds.
Analysis of thermodynamic formation quantities
The thermodynamic parameters namely enthalpy of formation ΔfHo (kcal/mol), entropy of formation ΔfSo (kcal/molK), and free enthalpy of formation ΔfGo (kcal/mol) were explored. It should be noted that a variation in enthalpy reflects the thermicity of a chemical reaction when that of the entropy provides information about the level of disorder in the system. On the other hand, a variation in free enthalpy reflects the spontaneity with which a chemical reaction occurs. These thermodynamic quantities in our study were obtained after optimization and frequency calculation, at the B3LYP/6-31+G (d, p) level. The values of the thermodynamic parameters are given in Table 1.
| Molecules |
|
|
|
|---|---|---|---|
| ITS 1 | -1219.335 | -0.935 | -940.438 |
| ITS 2 | -1219.502 | -0.935 | -940.673 |
| ITS 3 | -1219.081 | -0.935 | -940.322 |
| ITS 4 | -1177.678 | -0.933 | -899.421 |
| ITS 5 | -1177.422 | -0.933 | -899.105 |
| ITS 6 | -1178.215 | -0.933 | -899.954 |
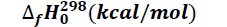
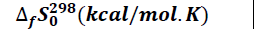
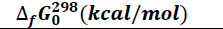
The results show that all the values of the standard thermodynamic quantities for molecule formation are negative. These negative values of enthalpy and free enthalpy reflect an exothermic and spontaneous reaction respectively under the conditions of the study. For entropy, a negative value indicates a decrease in disorder. Thus, the formation of all compounds is spontaneous with a release of heat and a decrease of disorder. At this level, we note that the determined quantities at the level of theory B3LYP/6-31+G (d, p) confirm the formation and the existence of the explored series of Imidazole-Thiosemicarbazides at required conditions of temperature and pressure (298.15K and 1 atm).
Global Descriptors
The study of the global chemical reactivity of molecules is based on the calculation of global indices deduced from the electronic properties. The global indices of the chemical reactivity of the studied ITSs are recorded in Table 2.
The results in Table 2 show that the ITS 1 compound has the smallest energy gap value (ΔE=4.630 eV), so this compound is more reactive and less stable. In contrast, the ITS 3 compound which has the largest energy gap (ΔE=4.744 eV), is therefore the less reactive and the more stable among studied molecules. Thus, we can establish the following sequence in decreasing order of reactivity:
| Molecules |
|
|
ΔE (eV) | χ(eV) | µ(eV) | ÃÂ? (eV) | ω(eV) | |
|---|---|---|---|---|---|---|---|---|
| ITS 1 | -6.331 | -1.701 | 4.630 | 4.016 | -4.016 | 2.315 | 3.483 | |
| ITS 2 | -6.416 | -1.738 | 4.678 | 4.077 | -4.077 | 2.339 | 3.553 | |
| ITS 3 | -6.38 | -1.636 | 4.744 | 4.008 | -4.008 | 2.372 | 3.387 | |
| ITS 4 | -6.349 | -1.716 | 4.633 | 4.032 | -4.032 | 2.317 | 3.510 | |
| ITS 5 | -6.434 | -1.738 | 4.696 | 4.086 | -4.086 | 2.348 | 3.555 | |
| ITS 6 | -6.37 | -1.728 | 4.643 | 4.049 | -4.049 | 2.321 | 3.531 |
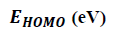
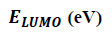
This decreasing order of stability is shown in Figure 2.
This decreasing order of stability is shown in Figure 2.

Figure 2. Representation of the energy gap of the imidazole-thiosemicarbazide series by increasing gap value.
The other interpreted parameter is chemical hardness (η). Compounds ITS 1 and ITS 4 have the lowest values (2.315 eV and 2.317 eV) respectively compared to the other compounds, indicating that they are the least hard (soft) of the studied compounds.
In summary, the overall descriptors revealed that ITS 1 and ITS 4 were the most reactive, least stable and softest compounds. On the other hand, ITS 1 was the softest of the studied compounds.
Local Descriptors
In the study of the isodensity map, a site is likely to be nucleophilic or electrophilic if it belongs to a larger lobe [27]. The isodensity maps showing the probable nucleophilic and electrophilic attack sites using the large lobes of the six (6) studied compounds are shown below in Figures 3-8.

Figure 3. HOMO and LUMO isodensity maps of the ITS 1 compound.

Figure 4. HOMO and LUMO isodensity maps of the ITS 2 compound.

Figure 5. HOMO and LUMO isodensity maps of the ITS 3 compound.

Figure 6. HOMO and LUMO isodensity maps of the ITS 4 compound.

Figure 7. HOMO and LUMO isodensity maps of the ITS 5 compound.

Figure 8. HOMO and LUMO isodensity maps of the ITS 6 compound.
Analysis of the maps through HOMO indicates that the largest lobe entirely containing the C28, C1 and N14 atoms would be the probable nucleophilic sites of the series of six (6) studied compounds. With respect to the electrophilic sites of attack obtained from LUMO; atoms C18, C12, and S21 have the largest lobes. These would appear to be the likely electrophilic sites of the studied series of halogenated ITSs.
In order to predict accurately the sites of electrophilic and nucleophilic attack, local and dual descriptors of reactivity were determined for each compound according to equations 7 to 15. Only heavy atoms are taken into account in this study. These different descriptors of reactivity are grouped in Tables 3-8.
| Local descriptors | Dual descriptors | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Atoms | f+ | f- | S+ | S- | ω+ | ω- | Δf(r) | ΔS(r) | ΔW(r) |
| C1 | 0.0908 | 0.0910 | 0.0388 | 0.0389 | 0.3225 | 0.3235 | -0.0003 | -0.0001 | -0.0009 |
| C2 | 0.0134 | 0.0366 | 0.0057 | 0.0157 | 0.0478 | 0.1301 | -0.0232 | -0.0099 | -0.0823 |
| C3 | 0.0388 | 0.0407 | 0.0166 | 0.0174 | 0.1379 | 0.1448 | -0.0019 | -0.0008 | -0.0068 |
| C6 | 0.0508 | 0.0531 | 0.0217 | 0.0227 | 0.1804 | 0.1885 | -0.0023 | -0.0010 | -0.0081 |
| N10 | 0.0337 | 0.0361 | 0.0144 | 0.0154 | 0.1198 | 0.1284 | -0.0024 | -0.0010 | -0.0086 |
| N11 | 0.0436 | 0.0344 | 0.0186 | 0.0147 | 0.1549 | 0.1223 | 0.0092 | 0.0039 | 0.0327 |
| C12 | 0.0625 | 0.0156 | 0.0267 | 0.0067 | 0.2219 | 0.0553 | 0.0469 | 0.0201 | 0.1667 |
| O13 | 0.0655 | 0.0386 | 0.0280 | 0.0165 | 0.2328 | 0.1373 | 0.0269 | 0.0115 | 0.0955 |
| N14 | 0.0234 | 0.0744 | 0.0100 | 0.0318 | 0.0831 | 0.2644 | -0.0510 | -0.0218 | -0.1813 |
| N16 | 0.0406 | 0.0833 | 0.0174 | 0.0356 | 0.1442 | 0.2958 | -0.0427 | -0.0182 | -0.1516 |
| C18 | 0.0687 | 0.0050 | 0.0294 | 0.0021 | 0.2440 | 0.0176 | 0.0637 | 0.0272 | 0.2264 |
| N19 | 0.0328 | 0.0701 | 0.0140 | 0.0300 | 0.1164 | 0.2490 | -0.0373 | -0.0160 | -0.1326 |
| S21 | 0.1480 | 0.0848 | 0.0633 | 0.0363 | 0.5257 | 0.3013 | 0.0632 | 0.0270 | 0.2244 |
| C22 | 0.0195 | 0.0283 | 0.0083 | 0.0121 | 0.0693 | 0.1006 | -0.0088 | -0.0038 | -0.0312 |
| C23 | 0.0209 | 0.0343 | 0.0089 | 0.0147 | 0.0743 | 0.1218 | -0.0134 | -0.0057 | -0.0475 |
| C24 | 0.0394 | 0.0434 | 0.0168 | 0.0185 | 0.1398 | 0.1541 | -0.0040 | -0.0017 | -0.0143 |
| C25 | 0.0575 | 0.0534 | 0.0246 | 0.0228 | 0.2043 | 0.1896 | 0.0041 | 0.0018 | 0.0146 |
| C26 | 0.0457 | 0.0566 | 0.0195 | 0.0242 | 0.1624 | 0.2011 | -0.0109 | -0.0047 | -0.0387 |
| C28 | 0.0860 | 0.0928 | 0.0367 | 0.0397 | 0.3054 | 0.3296 | -0.0068 | -0.0029 | -0.0242 |
| F32 | 0.0184 | 0.0275 | 0.0078 | 0.0117 | 0.0652 | 0.0976 | -0.0091 | -0.0039 | -0.0324 |
| Local descriptors | Dual descriptors | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Atoms | f+ | f- | S+ | S- | ω+ | ω- | Δf(r) | ΔS(r) | ΔW(r) |
| C1 | 0.0889 | 0.0939 | 0.0380 | 0.0401 | 0.3158 | 0.3335 | -0.0050 | -0.0021 | -0.0177 |
| C2 | 0.0128 | 0.0396 | 0.0055 | 0.0169 | 0.0456 | 0.1406 | -0.0268 | -0.0114 | -0.0951 |
| C3 | 0.0378 | 0.0423 | 0.0161 | 0.0181 | 0.1341 | 0.1503 | -0.0046 | -0.0019 | -0.0162 |
| C6 | 0.0494 | 0.0549 | 0.0211 | 0.0235 | 0.1755 | 0.1950 | -0.0055 | -0.0023 | -0.0195 |
| N10 | 0.0331 | 0.0371 | 0.0141 | 0.0159 | 0.1175 | 0.1318 | -0.0040 | -0.0017 | -0.0143 |
| N11 | 0.0425 | 0.0359 | 0.0182 | 0.0154 | 0.1508 | 0.1276 | 0.0065 | 0.0028 | 0.0233 |
| C12 | 0.0605 | 0.0155 | 0.0259 | 0.0066 | 0.2150 | 0.0551 | 0.0450 | 0.0192 | 0.1599 |
| O13 | 0.0633 | 0.0393 | 0.0271 | 0.0168 | 0.2248 | 0.1397 | 0.0240 | 0.0102 | 0.0852 |
| N14 | 0.0231 | 0.0745 | 0.0099 | 0.0319 | 0.0819 | 0.2647 | -0.0515 | -0.0220 | -0.1828 |
| N16 | 0.0417 | 0.0824 | 0.0178 | 0.0352 | 0.1480 | 0.2926 | -0.0407 | -0.0174 | -0.1446 |
| C18 | 0.0679 | 0.0057 | 0.0290 | 0.0025 | 0.2412 | 0.0204 | 0.0622 | 0.0266 | 0.2208 |
| N19 | 0.0452 | 0.0697 | 0.0193 | 0.0298 | 0.1604 | 0.2477 | -0.0246 | -0.0105 | -0.0873 |
| S21 | 0.1490 | 0.0886 | 0.0637 | 0.0379 | 0.5292 | 0.3146 | 0.0604 | 0.0258 | 0.2146 |
| C22 | 0.0198 | 0.0242 | 0.0084 | 0.0103 | 0.0702 | 0.0860 | -0.0044 | -0.0019 | -0.0157 |
| C23 | 0.0376 | 0.0370 | 0.0161 | 0.0158 | 0.1336 | 0.1316 | 0.0006 | 0.0002 | 0.0020 |
| C24 | 0.0456 | 0.0587 | 0.0195 | 0.0251 | 0.1621 | 0.2087 | -0.0131 | -0.0056 | -0.0466 |
| C25 | 0.0226 | 0.0265 | 0.0097 | 0.0113 | 0.0802 | 0.0941 | -0.0039 | -0.0017 | -0.0139 |
| C26 | 0.0564 | 0.0533 | 0.0241 | 0.0228 | 0.2003 | 0.1894 | 0.0031 | 0.0013 | 0.0109 |
| C28 | 0.0810 | 0.0921 | 0.0346 | 0.0394 | 0.2877 | 0.3272 | -0.0111 | -0.0047 | -0.0395 |
| F32 | 0.0220 | 0.0287 | 0.0094 | 0.0123 | 0.0781 | 0.1020 | -0.0067 | -0.0029 | -0.0239 |
| Local descriptors | Dual descriptors | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Atoms | f+ | f- | S+ | S- | ω+ | ω- | Δf(r) | ΔS(r) | ΔW(r) |
| C1 | 0.0931 | 0.0913 | 0.0393 | 0.0385 | 0.3153 | 0.3091 | 0.0018 | 0.0008 | 0.0062 |
| C2 | 0.0147 | 0.0380 | 0.0062 | 0.0160 | 0.0499 | 0.1287 | -0.0233 | -0.0098 | -0.0788 |
| C3 | 0.0397 | 0.0407 | 0.0167 | 0.0171 | 0.1345 | 0.1377 | -0.0009 | -0.0004 | -0.0032 |
| C6 | 0.0524 | 0.0528 | 0.0221 | 0.0223 | 0.1773 | 0.1790 | -0.0005 | -0.0002 | -0.0016 |
| N10 | 0.0344 | 0.0362 | 0.0145 | 0.0152 | 0.1165 | 0.1225 | -0.0018 | -0.0007 | -0.0060 |
| N11 | 0.0451 | 0.0350 | 0.0190 | 0.0148 | 0.1527 | 0.1186 | 0.0101 | 0.0042 | 0.0341 |
| C12 | 0.0629 | 0.0153 | 0.0265 | 0.0065 | 0.2131 | 0.0519 | 0.0476 | 0.0201 | 0.1612 |
| O13 | 0.0653 | 0.0388 | 0.0275 | 0.0163 | 0.2210 | 0.1313 | 0.0265 | 0.0112 | 0.0898 |
| N14 | 0.0254 | 0.0694 | 0.0107 | 0.0293 | 0.0862 | 0.2350 | -0.0440 | -0.0185 | -0.1489 |
| N16 | 0.0403 | 0.0788 | 0.0170 | 0.0332 | 0.1365 | 0.2667 | -0.0384 | -0.0162 | -0.1302 |
| C18 | 0.0652 | 0.0075 | 0.0275 | 0.0032 | 0.2209 | 0.0254 | 0.0577 | 0.0243 | 0.1955 |
| N19 | 0.0545 | 0.0699 | 0.0230 | 0.0295 | 0.1845 | 0.2368 | -0.0154 | -0.0065 | -0.0523 |
| S21 | 0.1453 | 0.1003 | 0.0613 | 0.0423 | 0.4921 | 0.3399 | 0.0450 | 0.0190 | 0.1523 |
| C22 | 0.0152 | 0.0283 | 0.0064 | 0.0119 | 0.0515 | 0.0960 | -0.0131 | -0.0055 | -0.0445 |
| C23 | 0.0276 | 0.0419 | 0.0116 | 0.0177 | 0.0935 | 0.1419 | -0.0143 | -0.0060 | -0.0484 |
| C24 | 0.0542 | 0.0491 | 0.0229 | 0.0207 | 0.1836 | 0.1664 | 0.0051 | 0.0021 | 0.0172 |
| C25 | 0.0464 | 0.0510 | 0.0195 | 0.0215 | 0.1570 | 0.1728 | -0.0047 | -0.0020 | -0.0158 |
| C26 | 0.0481 | 0.0611 | 0.0203 | 0.0258 | 0.1630 | 0.2069 | -0.0130 | -0.0055 | -0.0439 |
| C28 | 0.0398 | 0.0501 | 0.0168 | 0.0211 | 0.1349 | 0.1696 | -0.0102 | -0.0043 | -0.0347 |
| F31 | 0.0302 | 0.0444 | 0.0127 | 0.0187 | 0.1022 | 0.1505 | -0.0143 | -0.0060 | -0.0484 |
| Local descriptors | Dual descriptors | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Atoms | f+ | f- | S+ | S- | ω+ | ω- | Δf(r) | ΔS(r) | ΔW(r) |
| C1 | 0.0891 | 0.0900 | 0.0385 | 0.0389 | 0.3127 | 0.3160 | -0.0009 | -0.0004 | -0.0033 |
| C2 | 0.0129 | 0.0358 | 0.0056 | 0.0154 | 0.0454 | 0.1255 | -0.0228 | -0.0099 | -0.0801 |
| C3 | 0.0381 | 0.0401 | 0.0164 | 0.0173 | 0.1336 | 0.1408 | -0.0020 | -0.0009 | -0.0072 |
| C6 | 0.0494 | 0.0520 | 0.0213 | 0.0224 | 0.1735 | 0.1824 | -0.0026 | -0.0011 | -0.0090 |
| N10 | 0.0332 | 0.0358 | 0.0143 | 0.0154 | 0.1164 | 0.1255 | -0.0026 | -0.0011 | -0.0092 |
| N11 | 0.0427 | 0.0339 | 0.0184 | 0.0147 | 0.1498 | 0.1191 | 0.0087 | 0.0038 | 0.0306 |
| C12 | 0.0615 | 0.0155 | 0.0266 | 0.0067 | 0.2159 | 0.0545 | 0.0460 | 0.0199 | 0.1614 |
| O13 | 0.0644 | 0.0386 | 0.0278 | 0.0167 | 0.2261 | 0.1356 | 0.0258 | 0.0111 | 0.0905 |
| N14 | 0.0220 | 0.0736 | 0.0095 | 0.0318 | 0.0771 | 0.2583 | -0.0516 | -0.0223 | -0.1812 |
| N16 | 0.0402 | 0.0814 | 0.0174 | 0.0352 | 0.1412 | 0.2858 | -0.0412 | -0.0178 | -0.1447 |
| C18 | 0.0688 | 0.0053 | 0.0297 | 0.0023 | 0.2414 | 0.0186 | 0.0635 | 0.0274 | 0.2228 |
| N19 | 0.0308 | 0.0672 | 0.0133 | 0.0290 | 0.1081 | 0.2360 | -0.0364 | -0.0157 | -0.1279 |
| S21 | 0.1479 | 0.0884 | 0.0638 | 0.0382 | 0.5190 | 0.3104 | 0.0594 | 0.0257 | 0.2086 |
| C22 | 0.0204 | 0.0250 | 0.0088 | 0.0108 | 0.0716 | 0.0879 | -0.0046 | -0.0020 | -0.0162 |
| C23 | 0.0202 | 0.0282 | 0.0087 | 0.0122 | 0.0710 | 0.0991 | -0.0080 | -0.0034 | -0.0280 |
| C24 | 0.0335 | 0.0398 | 0.0145 | 0.0172 | 0.1176 | 0.1397 | -0.0063 | -0.0027 | -0.0221 |
| C25 | 0.0493 | 0.0489 | 0.0213 | 0.0211 | 0.1729 | 0.1716 | 0.0004 | 0.0002 | 0.0012 |
| C26 | 0.0465 | 0.0519 | 0.0201 | 0.0224 | 0.1630 | 0.1820 | -0.0054 | -0.0023 | -0.0190 |
| C28 | 0.0835 | 0.0879 | 0.0361 | 0.0380 | 0.2931 | 0.3085 | -0.0044 | -0.0019 | -0.0154 |
| Cl31 | 0.0455 | 0.0604 | 0.0196 | 0.0261 | 0.1595 | 0.2120 | -0.0150 | -0.0065 | -0.0525 |
| Local descriptors | Dual descriptors | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Atoms | f+ | f- | S+ | S- | ω+ | ω- | Δf(r) | ΔS(r) | ΔW(r) |
| C1 | 0.0863 | 0.0926 | 0.0368 | 0.0395 | 0.3070 | 0.3294 | -0.0063 | -0.0027 | -0.0224 |
| C2 | 0.0122 | 0.0394 | 0.0052 | 0.0168 | 0.0435 | 0.1400 | -0.0271 | -0.0116 | -0.0965 |
| C3 | 0.0366 | 0.0418 | 0.0156 | 0.0178 | 0.1301 | 0.1485 | -0.0052 | -0.0022 | -0.0184 |
| C6 | 0.0480 | 0.0542 | 0.0204 | 0.0231 | 0.1707 | 0.1928 | -0.0062 | -0.0026 | -0.0221 |
| N10 | 0.0322 | 0.0366 | 0.0137 | 0.0156 | 0.1145 | 0.1300 | -0.0044 | -0.0019 | -0.0155 |
| N11 | 0.0411 | 0.0355 | 0.0175 | 0.0151 | 0.1461 | 0.1262 | 0.0056 | 0.0024 | 0.0199 |
| C12 | 0.0585 | 0.0152 | 0.0249 | 0.0065 | 0.2078 | 0.0539 | 0.0433 | 0.0184 | 0.1539 |
| O13 | 0.0612 | 0.0385 | 0.0261 | 0.0164 | 0.2175 | 0.1370 | 0.0226 | 0.0096 | 0.0805 |
| N14 | 0.0226 | 0.0730 | 0.0096 | 0.0311 | 0.0805 | 0.2595 | -0.0504 | -0.0214 | -0.1790 |
| N16 | 0.0414 | 0.0807 | 0.0176 | 0.0344 | 0.1472 | 0.2869 | -0.0393 | -0.0167 | -0.1397 |
| C18 | 0.0663 | 0.0059 | 0.0282 | 0.0025 | 0.2356 | 0.0209 | 0.0604 | 0.0257 | 0.2148 |
| N19 | 0.0465 | 0.0680 | 0.0198 | 0.0290 | 0.1653 | 0.2418 | -0.0215 | -0.0092 | -0.0765 |
| S21 | 0.1458 | 0.0864 | 0.0621 | 0.0368 | 0.5183 | 0.3072 | 0.0594 | 0.0253 | 0.2112 |
| C22 | 0.0211 | 0.0232 | 0.0090 | 0.0099 | 0.0751 | 0.0825 | -0.0021 | -0.0009 | -0.0074 |
| C23 | 0.0299 | 0.0297 | 0.0127 | 0.0127 | 0.1062 | 0.1056 | 0.0001 | 0.0001 | 0.0005 |
| C24 | 0.0491 | 0.0574 | 0.0209 | 0.0244 | 0.1747 | 0.2040 | -0.0082 | -0.0035 | -0.0293 |
| C25 | 0.0192 | 0.0212 | 0.0082 | 0.0090 | 0.0684 | 0.0752 | -0.0019 | -0.0008 | -0.0069 |
| C26 | 0.0535 | 0.0510 | 0.0228 | 0.0217 | 0.1903 | 0.1814 | 0.0025 | 0.0011 | 0.0089 |
| C28 | 0.0802 | 0.0863 | 0.0342 | 0.0368 | 0.2852 | 0.3069 | -0.0061 | -0.0026 | -0.0217 |
| Cl32 | 0.0480 | 0.0634 | 0.0205 | 0.0270 | 0.1707 | 0.2255 | -0.0154 | -0.0066 | -0.0548 |
| Local descriptors | Dual descriptors | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Atoms | f+ | f- | S+ | S- | ω+ | ω- | Δf(r) | ΔS(r) | ΔW(r) |
| C1 | 0.0866 | 0.0854 | 0.0228 | 0.0225 | 0.2993 | 0.2950 | 0.0012 | 0.0003 | 0.0043 |
| C2 | 0.0125 | 0.0347 | 0.0033 | 0.0091 | 0.0432 | 0.1198 | -0.0222 | -0.0058 | -0.0766 |
| C3 | 0.0367 | 0.0378 | 0.0097 | 0.0099 | 0.1270 | 0.1305 | -0.0010 | -0.0003 | -0.0036 |
| C6 | 0.0481 | 0.0490 | 0.0127 | 0.0129 | 0.1664 | 0.1694 | -0.0009 | -0.0002 | -0.0031 |
| N10 | 0.0323 | 0.0338 | 0.0085 | 0.0089 | 0.1115 | 0.1169 | -0.0016 | -0.0004 | -0.0054 |
| N11 | 0.0413 | 0.0325 | 0.0109 | 0.0086 | 0.1428 | 0.1123 | 0.0088 | 0.0023 | 0.0306 |
| C12 | 0.0586 | 0.0144 | 0.0154 | 0.0038 | 0.2026 | 0.0498 | 0.0442 | 0.0116 | 0.1528 |
| O13 | 0.0612 | 0.0359 | 0.0161 | 0.0094 | 0.2116 | 0.1239 | 0.0254 | 0.0067 | 0.0876 |
| N14 | 0.0228 | 0.0649 | 0.0060 | 0.0171 | 0.0786 | 0.2244 | -0.0422 | -0.0111 | -0.1458 |
| N16 | 0.0406 | 0.0724 | 0.0107 | 0.0191 | 0.1404 | 0.2503 | -0.0318 | -0.0084 | -0.1099 |
| C18 | 0.0652 | 0.0058 | 0.0172 | 0.0015 | 0.2253 | 0.0200 | 0.0594 | 0.0156 | 0.2052 |
| N19 | 0.0473 | 0.0698 | 0.0124 | 0.0184 | 0.1634 | 0.2413 | -0.0225 | -0.0059 | -0.0778 |
| S21 | 0.1439 | 0.0874 | 0.0379 | 0.0230 | 0.4974 | 0.3020 | 0.0565 | 0.0149 | 0.1954 |
| C22 | 0.0198 | 0.0298 | 0.0052 | 0.0079 | 0.0684 | 0.1031 | -0.0101 | -0.0026 | -0.0347 |
| C23 | 0.0323 | 0.0435 | 0.0085 | 0.0115 | 0.1118 | 0.1505 | -0.0112 | -0.0029 | -0.0387 |
| C24 | 0.0515 | 0.0510 | 0.0136 | 0.0134 | 0.1780 | 0.1763 | 0.0005 | 0.0001 | 0.0017 |
| C25 | 0.0455 | 0.0476 | 0.0120 | 0.0125 | 0.1571 | 0.1646 | -0.0022 | -0.0006 | -0.0075 |
| C26 | 0.0497 | 0.0554 | 0.0131 | 0.0146 | 0.1717 | 0.1914 | -0.0057 | -0.0015 | -0.0197 |
| C28 | 0.0395 | 0.0451 | 0.0104 | 0.0119 | 0.1365 | 0.1558 | -0.0056 | -0.0015 | -0.0194 |
| Cl32 | 0.0645 | 0.1038 | 0.0170 | 0.0273 | 0.2228 | 0.3586 | -0.0393 | -0.0103 | -0.1359 |
Analysis of the local descriptors in Table 3 shows that the sulphur atom S21 is the nucleophilic site of attack and the carbon atom C28 is the electrophilic site of attack. Also, the analysis of the local descriptors in Table 4 indicates that the sulphur atom S21 is the nucleophilic site of attack but the carbon atom C1 becomes the electrophilic site of attack. In addition, Table 5 shows that the sulphur atom S21 is the nucleophilic and electrophilic site of attack.
The values of the local descriptors in Tables 6 and 7 show that the sulphur atom S21 is the nucleophilic site of attack and the carbon atom C1 is the electrophilic site of attack. Also, Table 8 shows that the sulphur atom S21 is the nucleophilic site of attack and the chlorine atom Cl32 is the electrophilic site of attack.
The observation made when analyzing the data in Tables 3 to 8 is that the results are mixed, for example Table 7 where the sulphur atom is both a nucleophilic and an electrophilic site. This result does not allow us to identify the electrophilic and nucleophilic sites. This leads us to the use of dual descriptors that will be used as ideal descriptors of the regioselectivity of the different sites of attack. The values of the dual descriptors of the series of halogenated imidazole-thiosemicarbazides, calculated at the B3LYP/6-31+G level (d, p), show that the nitrogen atom N14 is the preferred site of electrophilic attack. According to this same level of calculation, nucleophilic attack will preferentially take place on the C18 atom. These electrophilic and nucleophilic attack sites (N14 and C18) are identical for all compounds according to the dual descriptors. It is thus retained that the substitution of halogens on imidazole-thiosemicarbazides does not modify the centres of reactivity.
Hierarchical Ascending Classification Analysis (HAC)
The Ascending Hierarchical Classification (AHC) of ITSs was illustrated by the dendrogram in Figure 9. The purpose of the dendrogram is to partition a set of compounds into homogeneous groups or classes [28,29]. It assembles molecules by aggregation the molecules that are most similar to each other using measures of dissimilarity or distance between compounds to form classes. The horizontal lines represent the compounds. Vertical lines represent similarity values between pairs of compounds, a compound and a group of compounds, and among groups of compounds.

Figure 9. Dendrogram obtained for the studied ITSs.
From this analysis, we can see that the studied compounds have been grouped into three categories: the most active, which is ITS 3, the moderately active compounds ITS 2 and ITS 5, and the less active compounds ITS 1, ITS 4 and ITS 6.
Surface profiler
The surface profiler is a three-dimensional plot with one or more dependent variables represented by a readable surface. The surface plot from JMP Pro 13 software [30] of few global descriptors ΔE, ω, and η is shown in Figure 10.

Figure 10. Surface profile of ITSs from a few descriptors.
The analysis of the surface profiler shows a nearly smooth plane connecting the three (3) descriptors. This result shows the linearity between the descriptors of reactivity.
CONCLUSION
In this work, Quantum Chemistry and Molecular Modelling methods were used on six (6) molecules of the family of halogenated imidazole-thiosemicarbazides in order to study their chemical reactivity. This theoretical study was carried out using the DFT method with the B3LYP/6-31+G (d, p) level. Global and local descriptors were used to study the reactivity of different nucleophilic and electrophilic sites and their influence on molecular interaction in a qualitative and quantitative manner. The obtained descriptors could also provide more information and contribute to a better understanding of the electronic structure of halogenated imidazole-thiosemicarbazides. The analysis of the thermodynamic quantities of formation confirmed the formation and existence of the series of studied molecules. The HOMO isodensity maps showed that the largest lobe containing entirely C28, C1 and N14 atoms would be the probable nucleophilic sites, while the HOMO isodensity maps showing C18, C12 and S21 atoms have the largest lobes would appear to be the probable electrophilic sites of the series of halogenated studied ITSs. As for the overall descriptors; they revealed that ITS 1 and ITS 4 compounds were the most reactive, least stable and softest. On the other hand, ITS 1 was the softest among studied compounds. Analysis of the local descriptors confirmed the same obtained sites of electrophilic and nucleophilic attack. In order to determine accurately the different sites of attack, Fukui indices and dual descriptors were calculated from the Hirshfeld population charges. These showed that for the series of studied molecules, the nitrogen atom N14 is the preferred site of electrophilic attack and the carbon atom C18 is the preferred site of nucleophilic attack.
REFERENCES
- Paneth A, Węglińska L, Bekier A, et al., Molecules. 2019, 24: p. 1-14.
- Montoya J. Toxoplasmose. 2004, 363: p.1965-1976.
- MCauley J, Boyer KM, Patel D, et al., Clinical Infectious Diseases. 1994, 19: p. 38-72.
- Daffos F, Mirlesse V, Hohlfeld P, et al., Lancet. 1994, 344: p. 541.
- Kurt M, Sertbakan TR, Ozduran M. Acta Part A: Mol Biomol Spectrosc. 2008, 70: p. 664-673.
- Bohoussou KV, Benié A, Koné MG, et al., Computational Chemistry. 2017, 5: p. 113-128.
- Soro D, Ekou L, Koné MR, et al., EJERS. 2019, 4: p. 45-49.
- Lee C, Yang W. Physical Review Journals. 1988, B37: p. 785.
- Axel BD. Chem Phys. 1993, 98: p. 5648.
- Bédé AL, Assoma AB, Yapo KD, et al., Computational Chemistry, 2018, 6: p. 57-70.
- Frisch MJ, Trucks GW, Schlegel HB. Wallingford CT. 2009.
- Kapp J, Remko M, Schleyer R. JACS. 1996, 118: p. 5745-5751.
- Johnson BG, Gill PM. The Journal of Chemical Physics. 1993, 98: p. 5612-5626.
- Parr RG, Yang W. Annual Review Physical Chemistry. 1995, 46: p. 701-728.
- Coulibaly WK, dri JN, Koné MGR, et al., Computational Molecular Bioscience. 2019, 9: p. 49-62.
- Bohoussou KV, Bénié A, Koné GRM, et al., Modern Chemistry. 2019, 7: p. 38-44.
- Addinsoft, XLSTAT and Addinsoft are Registered Trademarks of Addinsoft. 2014, p. 1995-2014.
- Hirshfeld FL. Theor Chim Acta. 1977, 44: p. 129-138.
- Chase MW, Davies CA, Downey JR, JANAF Thermochemical Tables. J Phys Ref. 1985, 14: p. 1985.
- Hirshfeld FL. Theor Chim Acta, 1977, 44: p. 129-138.
- Koopmans T. Physica. 1934, 1: p. 104-113.
- Dheivamalar S, Sugi L, Ambigai k. 2016, p. 17-31.
- Ayers PW, Parr RG. J Am Chem Soc. 2000, 122: p. 2010-2018.
- Fukui K, Yonezawa T, Shingu H. J Chem Phys. 1952, 20: p. 722-725.
- Morell C, Grand A, Toro-Labbé. Chem Phys Lett. 2006, 425: p. 342–346.
- N’dri JS, Koné MR, Kodjo CG, et al., Chem Sci Int J. 2018, 22: p. 1-11.
- Hong LX, Ling CH, R-Zhou ZZ. Acta. 2015, 137: p. 321-327.
- Escofier B. Dunod. 2008, p. 318.
- Kpidi H, Yapo O, Koné MG, et al., Am J Environ. 2018, 6: p. 1-9.
- JMPPro13, Statistical Discovery, Scintilla: SAS institute Inc., 1998-2014.
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref



