Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 6
Synthesis and In vitro Antimycobacterialactivity of Some Novel Pyrrolo[1,2-A]Pyrazine Incorporated Indolizine Derivatives
Murali Krishna Kolli1*, Karuna Raman Padi1, Neha Singh1, Vinay Bharadwaj Tatipamula2 and Raveendra Reddy P1
1Department of Chemistry, Sri Krishnadevaraya University, Ananthapuramu – 515003, Andhra Pradesh, India
2Pharmaceutical Chemistry Department, University College of Pharmaceutical Sciences, Andhra University, Visakhapatnam – 530003, Andhra Pradesh, India
- *Corresponding Author:
- Murali Krishna Kolli
Department of Chemistry Sri Krishnadevaraya University
Ananthapuramu – 515003, Andhra Pradesh, India
Abstract
The accumulating pharmacological importance of drug resistant pathogens has lent auxiliary urgency to new anti-tubercular compound development. In this regard, a novel series of pyrrolo [1,2-a]pyrazine incorporated indolizine derivatives have been synthesized, characterized by using spectral data and screened for in vitro antimycobacterial activity. The anti-tubercular activity of the synthesized compounds (7a-p) was determined by microplate alamar blue assay and the outcomes were screened against Mycobacterium tuberculosis H37Rv strain. Compounds 7a-p exhibited good to potent anti-tubercular activity when compared with the standard first line anti-tuberculosis drugs (ciprofloxacin, pyrazinamide and streptomycin). Some of the tested compounds exhibited highest inhibitory activity at 1.6 μg/ml minimal inhibition concentration.
Keywords
Indolizine, Mycobacterium tuberculosis, Antimycobacterial activity, MABA, Minimal inhibition concentration.
Introduction
Tuberculosis (TB) is an intercellular bacterium that vitiates, continues and replicates in resistant cells, which is endemic in distinct provinces of the biosphere and follows to be a leading worldwide health problem [1]. During the cure of TB, the Mycobacterium Tuberculosis undertakes mutational modifications and grows drug resistant bacterium which contributes to the elevated morbidity and mortality [2]. Therefore, the drug discovery of newer anti-TB agents with safe and potent biological action is required. This necessity had an impact on the synthesis for some novel indolizines with the anti-tubercular activity.
Indolizine derivative which are isosteric analogues of indolesare proved to exhibit various biological activity. These can be considered as privileged structures since they possess 30 different biological activities in the MDRR™ database including G-protein coupled receptors (GPCR) binding and modulation of DNA-protein interactions [3], some naturally occurring indolizines shown in Figure 1. In addition to natural products, synthetic indolizines also possess many biological activities such as anti-tubercular [4], anti-cancer [5], herbicidal [6], anti-histaminic [7], anti-inflammatory [8], aromatase [9] and phosphodiesterase [10] inhibitors. Many amino acid derivatives with an active indolizine nucleus have been utilized in cancer therapy [11]. These properties make indolizines much more attractive for both medicinal and synthetic chemists.
Figure 1: Naturally occurring indolizines
The pyrrolo[1,2-a]pyrazine are nitrogen rich heterocyclics that are known to appear in bioactive molecules, including serotonin receptor agonist, kappa opioid receptor antagonist, HCV inhibitors. A bibliographical search for the pyrrolo[1,2-a]quinoxalines revealed surprisingly that only a few synthetic procedures have been reported previously for these heterocyclic analogues, i.e. reduction of the lactam derivatives [12], 1,3-dipolar cycloaddition of quinoxalium N-ylides to alkenes [13] and alkynes [14] or reaction of 1-(2-aminophenyl)pyrroles with aldehydes [15]. As part of a program aimed at the discovery of novel polycyclic compounds with therapeutic potential (S)-1-(2-(4-methoxy-phenyl)-indolizin-3-yl)-2- (2,3,3a,4-tetra-hydro-pyrrolo[1,2-a]-quinoxalin-5(1H)-yl)ethane-1,2-dione has been and their biological activities has been evaluated.
Materials and Methods
Chemistry
The solvents were purified according to standard procedures prior to use, and all commercial chemicals were used as received. For Thin Layer chromatography (TLC) analysis, Merck pre-coated plates (silica gel 60 F254) were used and eluting solvents are indicated in the procedures. Merck silica gel 60 (230-400 mesh) was used for flash column chromatography. Melting point (mp) determinations were performed by using Mel-temp apparatus and are uncorrected. 1H-NMR spectra were recorded on a Varian Unity instrument at room temperature at 400 MHz. Chemical shifts are reported in ‘δ’ parts per million (ppm) downfield from tetramethylsilane (TMS) with reference to internal solvent and coupling constants ‘J’ in Hz. The mass spectra were recorded on Agilent ion trap MS. All the acid chlorides used for the preparation of 7a-q were purchased from commercial sources.
General procedure for the synthesis of 2-(4-methoxyphenyl)indolizine (2)
2-picoline (10 g, 107.5 mmol), 4-methoxy phenacyl bromide (24.6 g, 107.5 mmol) were dissolved in acetone and heated at reflux for 4 h. Precipated solid was filtered out and suspended in aq. NaHCO3 (200 mL) solution and heated at 90˚C for 9 h. Obtained solid was filtered and dried to get white solid 2.
Yield: 62.5%; M. p.: 80-95°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=8.72 (s, 1H), 8.54 (dd, J = 5.4 Hz, 1H), 7.65 (dd, J = 8.2 Hz, 1H), 7.46 (dd, J = 8.9 Hz, 2H), 7.19 (dd, J =8.9 Hz, 1H), 7.21 (dd, J = 6.9 Hz, 1H), 7.04 (dd, J = 1.7 Hz, 1H), 3.71 (s, 3H); ESI-MS: m/z, 224 (M+1).
General procedure for the synthesis of 2-(2-(4-methoxyphenyl)indolizin-3-yl)-2-oxoacetyl chloride (3)
1 (15 g, 67.26 mmol) was dissolved in 50 ml of toulene: THF (8: 2) and added oxalylchloride (8.4 g, 67.26 mmol) at 0°C. Reaction mixture was stirred at 0°C 2 h. After the completion of reaction solvent was removed under vacuum to get solid which was stirred in n-hexane and filtered. Obtained solid was dried under vacuum to get yellow solid 3.
Synthesis of [1-(3-nitropyridin-2-yl)pyrrolidin-2-yl]methanol (5)
To a stirred solution of 2-R(+)-hydroxymethyl pyrrolidine (6.39 g, 63.29 mmol), K2CO3 (10.5 g, 75.95 mmol) in dimethyl formamide added 2- chloro-3-nitropyridine (10 g, 63.29 mmol) at RT and stirred at room temperature for 4 h. After the completion of the reaction mass was poured into cold water and extracted with ethyl acetate. Organic layer was washed with water, brine, dried over Na2SO4 and evaporated under vacuum to get crude which was purified by column chromatography over Silica gel (#60-120) using n-hexane and ethyl acetate as eluent to get yellow solid 5.
Yield: 56.7 %;1H-NMR (400 MHz, DMSO-d6) δ (ppm)=8.30 (dd, J = 7.0 Hz 1H), 8.22 (dd, J = 5.2 Hz, 1H), 6.9 (dd, J = 7.0 Hz 1H), 3.91 (d, 2H), 3.77-3.86 (m, 2H), 3.45 (m, 1H), 2.1(m, 2H), 1.6-1.8 (m, 2H); ESI-MS: m/z, 224 (M+1).
Synthesis of [1-(3-aminopyridin-2-yl)pyrrolidin-2-yl]methanol (6)
5 (8 g, 35.87 mmol) was dissolved in methanol and hydrogenated over Pd/C (cat.) for 1 h. After completion of reaction catalyst was filtered and filtrate was concentrated to get brown solid 6.
Yield: 79.7%; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=7.8 (dd, J = 4.8 Hz 1H), 7.32 (dd, J = 8.3 Hz, 1H), 7.01 (dd, J = 8.3 Hz 1H), 3.85 (d, 2H), 3.7 (m, 1H), 3.3-3.5 (t, 2H), 1.8-2.1(m, 2H), 1.6-1.7(m, 2H); ESI-MS: m/z, 194 (M+1).
Synthesis of 5,6,6a,7,8,9-hexahydropyrido[3,2-e]pyrrolo[1,2-a]pyrazine (7)
6 (5 g, 35.87 mmol) was suspended in polyphosphoric acid (10 ml) and heated at 100°C for 1 h. Reaction mixture was cooled and added ice cold water, pH was adjusted to 8 using NaHCO3 and extracted with ethyl acetate, combined organic layer was washed with brine, dried over Na2SO4 and evaporated under vacuum to get crude. Purification by column using ethyl acetate as eluent gives 7 as brown solid.
Yield: 66.6%; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=7.35-7.33 (dd, J = 4.36 Hz 1H), 6.59-6.61 (dd, J = 8.36 Hz, 1H), 6.32-6.39 (m, 1H), 3.31-3.38 (m, 4H), 3.3-3.5 (m, 5H), 2.62-2.57 (m, 1H), 2.50-2.49(m, 1H), 2.05-2.02 (m, 1H), 1.95-1.85 (m, 2H); ESI-MS: m/z, 176 (M+1).
Synthesis of (R)-1-(2-(4-methoxyphenyl)indolizin-3-yl)-2-(6a,7,8,9-tetrahydropyrido[3,2-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)ethane-1,2-dione (7a)
7 (4.5 g, 25.7 mmol) was dissolved in THF added trimethylamine (3.9 g, 38.57 mmol), 3 (8 g, 25.71 mmol) at 0°C. Reaction mixture was stirred at RT for 1 h, after completion of reaction mass was concentrated to dryness added water, extracted with ethyl acetate, combined organic layer was washed with brine, dried over Na2SO4, concentrated to get crude, which was purified by column using ethyl acetate/hexane as eluent to get yellow solid as 7a.
Yield: 68.84%; M. p.: 242-246°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=9.89-9.88 (d, 1H), 7.85-7.82 (d, 2H), 7.72 (s, 1H), 7.53-7.49 (m, 2H), 7.34-7.32 (m, 1H), 6.91-6.86 (m, 3H), 6.72 (s, 1H), 6.37 (s, 1H), 4.21-4.18 (d, 1H), 3.81 (s, 1H), 3.66 (m, 3H), 3.52-3.49 (m, 2H), 2.71 (t, 1H), 2.08-1.96 (m, 1H), 1.89-1.73 (m, 2H); 1.44-1.29 (m, 1H); ESI-MS: m/z, 453 (M+1).
Synthesis of (R)-1-(2-(4-hydroxyphenyl)indolizin-3-yl)-2-(6a,7,8,9-tetrahydropyrido[3,2-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)ethane-1,2-dione (7b)
To an ice cooled solution of 7a (8 g, 17.69 mmol) in MDC, added borontribromide (11.4 g, 44.2 mmol) and stirred at 0˚C for 1 h. Reaction mixture was quenched into ice cold water, neutralized with aqueous ammonia, extracted with MDC. Combined organic layer was concentrated to get crude which was purified by column using ethyl acetate/hexane as eluent to get 7b as pale yellow solid.
Yield: 83.8%; M. p.: 240-245°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=9.89-9.88 (d, 1H), 7.85-7.82 (d, 2H), 7.72 (s, 1H), 7.53-7.49 (m, 2H), 7.34-7.32 (dd, 1H), 6.91-6.86 (m, 3H), 6.72 (s, 1H), 6.37 (s, 1H), 4.21-4.18 (d, 1H), 3.81 (s, 1H), 3.52-3.49 (m, 2H), 2.71 (t, 1H), 2.08-1.96 (m, 1H), 1.89-1.73 (m, 2H); 1.44-1.29 (m, 1H); 13C-NMR(400 MHz, DMSO-d6) δ (ppm)=178.5, 164.5, 158.0, 142.0, 139.3, 131.6, 131.2 (2C), 127.5, 126.8, 124.8, 118.9, 118.0, 117.8, 115.8, 115.1(2C), 110.5, 106.8, 106.5, 67.7, 47.1, 29.1, 23.5, 23.2; ESI-MS: m/z, 439 (M+1).
General experimental procedure for the preparation of ether derivatives (7c-q)
To a stirred solution of 7b, K2CO3 in DMF, added alkyl halides at RT and reaction mass was stirred at 50˚C for 4 h. After completion of reaction, added ice cold water and extracted with ethyl acetate. Combined organic washed with brine, dried over Na2SO4, concentrated to get crude, which was purified by column over silica gel using ethyl acetate/hexane as eluent to get 7c-q.
Synthesis of (R)-1-(2-(4-ethoxyphenyl)indolizin-3-yl)-2-(6a,7,8,9-tetrahydropyrido[3,2-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)ethane-1,2-dione (7c)
Pale yellow solid; yield: 59.3%; M. p.: 153- 158°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=9.89-9.87 (d, 1H), 7.85-7.78 (d, 2H), 7.65 (s, 1H), 7.53-7.48 (m, 2H), 7.32-7.30 (dd, 1H), 6.91-6.86 (m, 3H), 6.72 (s, 1H), 6.38-6.36 (s, 1H), 4.21-4.18 (d, 1H), 3.81 (s, 1H), 3.66 (m, 2H), 3.52-3.49 (m, 2H), 2.71 (t, 1H), 2.08-1.96 (m, 1H), 1.89-1.73 (m, 2H); 1.46-1.26 (m, 3H); 13C-NMR (400 MHz, DMSO-d6) δ (ppm)=178.4, 159.0, 148.4, 145.4, 144.7, 141.6, 139.3, 131.3(2C), 127.5, 126.4, 119.2, 118.1, 117.7, 115.9, 114.1(2C), 113.7, 110.2, 107.0, 106.6, 63.5, 47.1, 29.9, 23.5, 15.1, 15.0; ESI-MS: m/z, 467(M+1).
Synthesis of (R)-1-(2-(4-propoxyphenyl)indolizin-3-yl)-2-(6a,7,8,9-tetrahydropyrido[3,2-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)ethane-1,2-dione (7d)
Pale yellow solid; yield: 52.9%; M. p.: 162-166°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=10.06-10.00 (m, 1H), 7.85-7.82 (d, 2H), 7.77 (s, 1H), 7.57-7.46 (m, 2H), 7.34-7.32 (m, 1H), 7.09-7.02 (m, 3H), 6.82 (s, 1H), 6.37 (s, 1H), 4.21-4.18 (m, 1H), 3.81 (s, 1H), 3.52-3.49 (m, 2H), 2.71 (t, 1H), 2.08-1.96 (m, 1H), 1.89-1.73 (m, 2H); 1.441.29 (m, 1H), 1.14 (m, 1H), 0.89 (m, 2H), 0.64 (m, 2H); ESI-MS: m/z, 493 (M+1).
Synthesis of (R)-1-(2-(4-(2-morpholinoethoxy)phenyl)indolizin-3-yl)-2-(6a,7,8,9-tetrahydropyrido [3,2-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)ethane-1,2-dione (7e)
Yellow solid; yield: 62.3%; M. p.: 145-152°C, 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=9.88 (s, 1H), 7.82-7.82 (d, 2H), 7.49 (s, 1H), 7.31(m, 2H), 7.23 (dd, 1H), 6.90 (m, 3H), 6.72 (s, 1H), 6.55 (s, 1H), 4.21-4.18 (t, 2H), 3.94 (m, 5H), 3.60 (m, 4H), 3.34 (m, 4H), 2.50 (t, 4H), 2.08-1.96 (m, 4H);13C-NMR (400 MHz, DMSO-d6) δ (ppm)=178.4, 164.3, 158.8, 148.4, 145.4, 144.7, 141.5, 139.3, 131.6, 131.3(2C), 128.9, 127.5, 126.6, 119.2, 118.1, 117.7, 115.9, 114.2 (2C), 110.3, 107.0, 99.9, 66.5, 65.6, 57.2, 53.9, 53.8, 47.0, 29.9, 23.4; ESI-MS: m/z, 552 (M+1).
Synthesis of (R)-1-(2-(4-(2-morpholino-2-oxoethoxy)-phenyl)-indolizin-3-yl)-2-(6a,7,8,9-tetra-hydropyrido[3,2-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)-ethane-1,2-dione (7f)
Pale yellow solid; yield: 53.9%; M. p.: 145-152°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=9.99 (s, 1H), 7.87 (d, 1H), 7.77 (s, 1H), 7.60-7.58 (m, 2H), 7.48-7.27 (dd, 3H), 6.86 (m, 2H), 6.54-6.39 (m, 2H), 4.81(s, 2H), 4.54-4.72 (m, 1H), 4.16 (m, 1H), 3.64-3.28 (m, 10H), 2.40-2.37 (m, 2H), 2.06-1.97 (m, 2H); 13C-NMR (400 MHz, DMSO-d6) δ (ppm)=178.1, 166.2, 166.0, 158.0, 157.9, 147.9, 144.7, 144.0, 141.3, 139.4, 131.7, 131.2, 129.9, 127.7, 126.6, 118.4, 114.9, 114.0, 110.4, 106.6, 67.5, 66.2, 47.5, 46.6, 42.5, 30.1, 23.5; ESI-MS: m/z, 466 (M+1).
Synthesis of (R)-1-(2-(4-(2-phenoxyethoxy)phenyl)indolizin-3-yl)-2-(6a,7,8,9-tetrahydro pyrido [3,2-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)ethane-1,2-dione (7g)
Pale yellow solid; yield: 69.2%; M. p.: 124-128°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=10.01-9.99 (d, 1H), 7.86-7.85 (m, 2H), 7.60-7.58 (d, 1H), 7.35-7.27 (m, 4H), 7.12-7.10 (m, 2H), 7.04-6.95 (m, 4H), 6.91-6.86 (t, 2H), 6.56 (s, 1H), 4.36-4.28 (m, 4H), 4.22-4.16 (m, 5H), 2.05-1.99 (m, 2H), 1.97-1.90 (m, 2H); 13C-NMR (400 MHz, DMSO-d6) δ (ppm)=178.3, 164.4, 158.8, 158.2, 148.1, 145.2, 144.6, 141.6, 140.7, 139.2, 132.4, 131.6, 130.9, 129.8, 129.3(2C), 127.4, 126.7, 121.1, 118.5, 117.8, 114.7(2C). 110.2, 107.0, 66.3, 56.7, 47.6, 41.1, 36.7, 30.0, 23.4; ESI-MS: m/z, 560 (M+1).
Synthesis of (R)-1-(2-(4-(pentyloxy)phenyl)-indolizin-3-yl)-2-(6a,7,8,9-tetra-hydro-pyrido [3,2-e]-pyrrolo[1,2-a]-pyrazin-5(6H)-yl)ethane-1,2-dione (7h)
Pale yellow solid; yield: 52.3%; M. p.: 100-112°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=9.89-9.87 (d, 1H), 7.83-7.78 (d, 2H), 7.53-7.32 (m, 6H), 6.86 (m, 3H), 4.21-4.18 (d, 3H), 3.78 (m, 4H), 3.36 (m, 2H), 1.96 (m, 2H), 1.67 (m, 4H), 1.35 (m, 4H), 0.94 (t, 3H); ESI-MS: m/z, 509(M+1).
Synthesis of (R)-4-(3-(2-oxo-2-(6a,7,8,9-tetra-hydro-pyrido[3,2-e]pyrrolo[1,2-a]-pyrazin-5(6H)-yl)acetyl)-indolizin-2-yl)phenyl methane-sulfonate (7i)
Pale yellow solid; yield: 32.9%; M. p.: 144-156°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=10.06-9.99 (d, 1H), 7.83-7.82 (d, 2H), 7.75 (s, 1H), 7.58-7.49 (m, 2H), 7.35-7.27 (dd, 1H), 7.16-7.08 (m, 3H), 6.93 (s, 1H), 6.83-6.68 (s, 1H), 4.17 (s, 1H), 3.84-3.66 (m, 4H), 3.21 (t, 3H), 2.09-1.97 (m, 2H), 1.78 (m, 2H); 13C-NMR (400 MHz, DMSO-d6) δ (ppm)=157.5, 148.0, 143.8, 141.2, 139.4, 131.4, 131.1, 130.3, 129.4(2C), 126.8, 121.4, 118.5, 118.3, 118.2, 115.3, 114.8(2C), 114.9, 111.0, 106.8, 76.7, 47.6, 46.4, 30.0, 23.6, 23.5; ESI-MS: m/z, 517 (M+1).
Synthesis of (R)-ethyl 2-(4-(3-(2-oxo-2-(6a,7,8,9-tetrahydropyrido[3,2-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)acetyl)indolizin-2-yl)phenoxy)acetate (7j)
Pale yellow solid; yield: 59.5%; M. p.: 142-146°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=9.88 (d, 1H), 7.87-7.82 (d, 2H), 7.69 (s, 1H), 7.59 (m, 2H), 7.35-7.27 (dd, 1H), 7.27 (m, 3H), 7.12 (s, 1H), 6.83 (s, 1H), 4.63 (s, 2H), 4.17 (s, 2H), 3.67-3.55 (m, 5H), 2.06-1.97 (m, 4H), 0.60-0.85 (m, 4H); ESI-MS: m/z, 525(M+1).
Synthesis of (R)-N-cyclo-propyl-2-(4-(3-(2-oxo-2-(6a,7,8,9-tetra-hydro-pyrido[3,2-e]pyrrolo [1,2-a]-pyrazin-5-(6H)-yl)-acetyl)-indolizin-2-yl)phenoxy)-acetamide (7k)
Pale yellow solid; yield: 48.9%; M. p.: 142-148°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=10.00 (d, 1H), 7.87-7.82 (d, 2H), 7.79 (s, 1H), 7.59 (m, 2H), 7.35-7.27 (dd, 1H), 7.27 (m, 3H), 7.12 (s, 1H), 6.83 (s, 1H), 4.63 (s, 3H), 4.17 (s, 1H), 3.67-3.55 (m, 4H), 2.78 (t, 1H), 2.06-1.97 (m, 4H), 0.60-0.85 (m, 4H); ESI-MS: m/z, 536 (M+1).
Synthesis of (R)-1-(2-(4-(isopentyloxy)phenyl)-indolizin-3-yl)-2-(6a,7,8,9-tetra hydro pyrido [3,2-e]-pyrrolo[1,2-a]pyrazin-5(6H)-yl)ethane-1,2-dione (7l)
Pale yellow solid; yield: 66.2%; M. p.: 156-162°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=10.06-10.00 (d, 1H), 7.88-7.87 (d, 1H), 7.60-7.53 (d, 1H), 7.47 (s, 1H), 7.34-7.32 (m, 1H), 7.30-7.27 (m, 1H), 7.11-7.01 (m, 1H), 6.84-6.80 (m, 1H), 6.69 (s, 1H), 6.55 (s, 1H), 6.38 (s, 1H), 4.18 (d, 2H), 3.88 (m, 1H), 3.57-3.45 (m, 4H), 2.06-1.80 (m, 7H), 1.00- 0.94 (m, 6H); ESI-MS: m/z, 510 (M+1).
Synthesis of (R)-2-(4-(3-(2-oxo-2-(6a,7,8,9-tetrahydro-pyrido[3,2-e]pyrrolo[1,2-a] pyrazine-5(6H)-yl)acetyl)-indolizin-2-yl)phenoxy)-N-phenyl-acetamide (7m)
Pale yellow solid; yield: 59.3%, M. p.: 104-109°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=10.13-10.06 (d, 1H), 7.79 (d, 2H), 7.65 (d, 1H), 7.51 (m, 5H), 7.34-7.22 (m, 4H), 7.09 (s, 1H), 6.96 (s, 2H), 6.72 (s, 2H), 6.41 (s, 1H), 4.77 (s, 2H), 4.19 (s, 1H), 3.60-3.34 (m, 4H), 2.08-1.95 (m, 4H); ESI-MS: m/z, 572 (M+1).
Synthesis of (R)-N-(4-fluorophenyl)-2-(4-(3-(2-oxo-2-(6a,7,8,9-tetrahydropyrido[3,2-e]pyrrolo[1,2-a]pyrazin-5(6H)-yl)acetyl)indolizin-2-yl)phenoxy)acetamide (7n)
Pale yellow solid; yield: 52.3%; M. p.: 114-118°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=10.21-10.13 (d, 1H), 9.89 (s, 1H), 7.83-7.79 (m, 1H), 7.67 (m, 3H), 7.51 (m, 1H), 7.36 (s, 2H), 7.18 (s, 3H), 6.95 (m, 3H), 6.721 (s, 1H), 6.40 (m, 1H), 4.77 (m, 1H), 4.59 (m, 1H), 4.40 (m, 1H), 4.19 (m, 1H), 3.62 (m, 1H), 3.52 (m, 1H), 2.08 (m, 2H), 1.90 (m, 2H); ESI-MS: m/z, 590 (M+1).
Synthesis of (R)-1-(2-(4-(benzyloxy)-phenyl)-indolizin-3-yl)-2-(6a,7,8,9-tetra-hydro-pyrido [3,2-e]-pyrrolo[1,2-a]pyrazin-5(6H)-yl)ethane-1,2-dione (7o)
Pale yellow solid; yield: 63.5%; M. p.: 144-151°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=9.88-9.87 (d, 1H), 8.61 (s, 1H), 7.99-7.85 (m, 2H), 7.67 (m, 2H), 7.53-7.47 (m, 7H), 7.39-7.32 (m, 1H), 6.98-6.95 (m, 3H), 6.721 (s, 1H), 5.15 (s, 2H), 4.41 (m, 1H), 3.87 (m, 4H), 2.08 (m, 2H), 1.96 (m, 2H); ESI-MS: m/z, 529 (M+1).
Synthesis of 1-(2-(4-((2-oxooxazolidin-5-yl)methoxy)phenyl)indolizin-3-yl)-2-((R)-2,3,3a,4-tetra-hydro-pyrrolo[1,2-a]quinoxalin-5(1H)-yl)-ethane-1,2-dione (7p)
Pale yellow solid; yield: 39.5%; M. p.: 132-141°C; 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=9.89 (s, 1H), 7.81-7.63 (m, 4H), 7.53-7.47 (m, 4H), 6.92-6.73 (m, 4H), 6.39 (s, 1H), 4.87 (s, 1H), 4.20-3.91 (m, 4H), 3.62-3.33 (m, 5H), 2.08 (m, 2H), 1.96 (m, 2H); ESI-MS: m/z, 538 (M+1).
Antimycobacterial activity
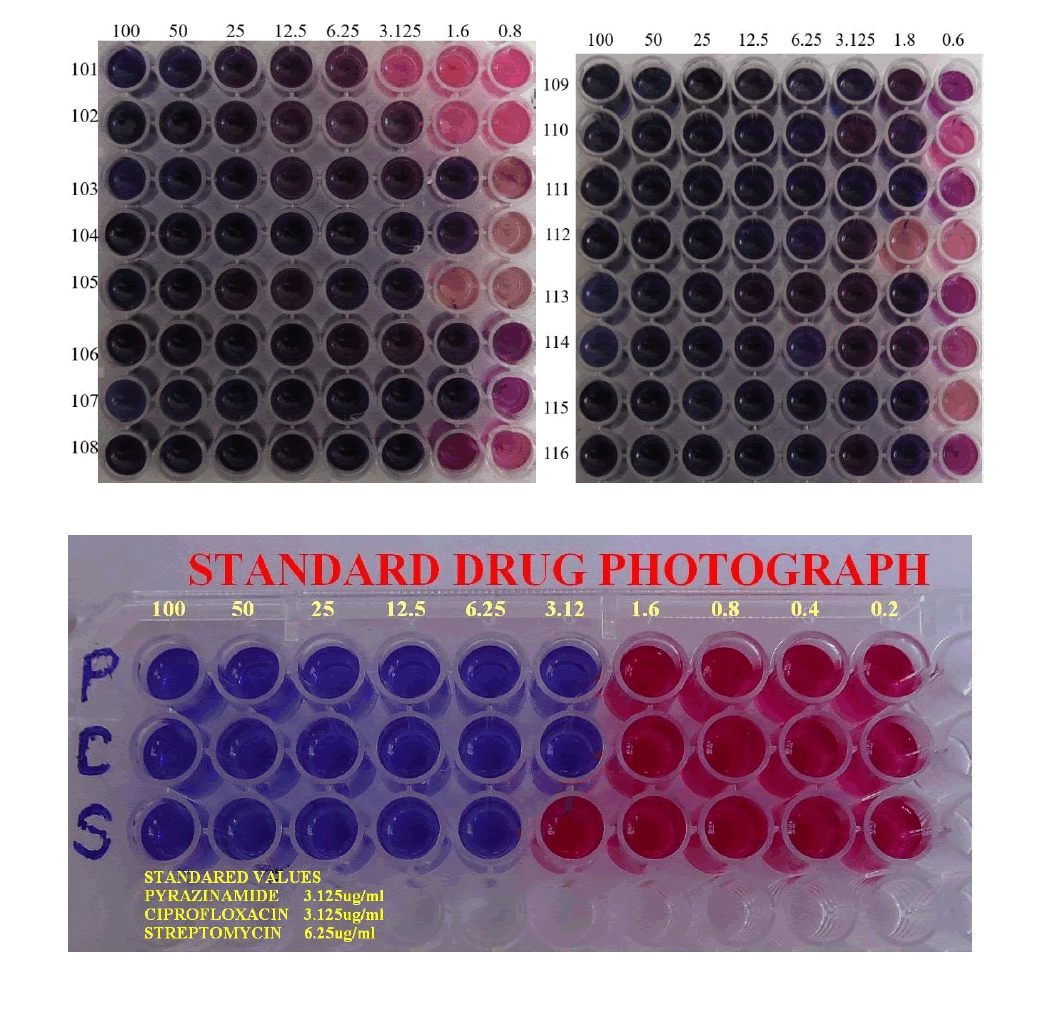
The in vitro antimycobacterial assessment of 7a-p have been tested against M. tuberculosis H37Rv strain ATCC 27294 using the micro plate alamar blue assay (MABA) and ciprofloxacin, pyrazinamide, streptomycin as standard drugs [16]. All the compounds 7a-p are screened for anti-tubercular activity in three sets (n=3). Initially, to lessen the evaporation of the medium in the aseptic 96 well plates during incubation, 200 μl of sterile deionized water was placed on all outer circumference wells of plate. Then to each well, 100 μl of the Middlebrook 7H9 broth was added and serial dilutions of the compounds 7a-p were made directly on the plate (i.e., 100-0.8 μg/ml). Then the 96 well plate was accurately occluded with parafilm and incubated at 37°C for five days. Finally, 25 μl of freshly prepared combination of almar blue (Accumed International, Westlake Ohio) reagent and 10% tween 80 (1: 1) were added to the well plate and incubated for one day. A blue and pink colour was elucidated as no bacterial growth and growth respectively. The Minimal Inhibition Concentration (MIC) is the lowest drug concentration, which averted a colour change from blue to pink.
Results and Discussion
Several methods has been disclosed for quinoxalines,like reductive cyclisation of nitro aldehydes using Ni-Raney in methanol which gives hexa-hydropyrrolo-quinoxaline in 76% yield. In the present invention we synthesised (S)-5,6,6a,7,8,9-hexahydropyrido[3,2-e]pyrrolo[1,2-a]pyrazinevia acid catalysed dehydration of amino alcohols derivatives and were linked with indolizine toproduce keto amide. The methoxy derivative which was demethylated using BBr3 to get hydroxy which is alkylated to get alkoxy derivatives. The entire synthetic scheme was depicted in Scheme 1.
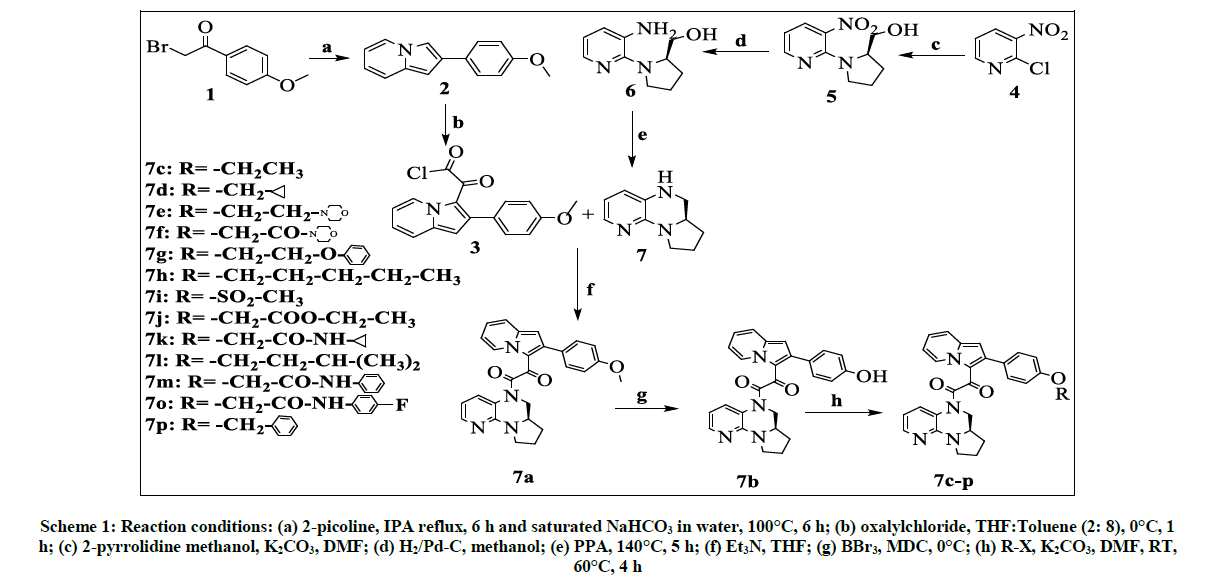
Scheme 1: Reaction conditions: (a) 2-picoline, IPA reflux, 6 h and saturated NaHCO3 in water, 100°C, 6 h; (b) oxalylchloride, THF:Toluene (2: 8), 0°C, 1 h; (c) 2-pyrrolidine methanol, K2CO3, DMF; (d) H2/Pd-C, methanol; (e) PPA, 140°C, 5 h; (f) Et3N, THF; (g) BBr3, MDC, 0°C; (h) R-X, K2CO3, DMF, RT, 60°C, 4 h
All the synthesized compounds were evaluated for in vitro anti-tubercular against M. tuberculosis and the results were tabulated in Table 1. Compounds 7c, 7d, 7f, 7g, 7i, 7j and 7k were very potent against M. tuberculosis H37Rv strain at MIC 1.6 μg/ml. Compounds 7b, 7e, 7h and 7l showed equivalent activity as that of standard drugs (pyrazinamide, ciprofloxacin) with a MIC 3.125 μg/ml. The compound 7a was found to be active against M. tuberculosis strainat MIC of 6.25 μg/ml similar to MIC of standard drug streptomycin.
| Compound | MIC values (µg/ml) |
|---|---|
| 7a | 6.25 |
| 7b | 3.125 |
| 7c | 1.6 |
| 7d | 1.6 |
| 7e | 3.125 |
| 7f | 1.6 |
| 7g | 1.6 |
| 7h | 3.125 |
| 7i | 1.6 |
| 7j | 1.6 |
| 7k | 1.6 |
| 7l | 3.125 |
| 7m | 1.6 |
| 7n | 1.6 |
| 7o | 1.6 |
| 7p | 1.6 |
| Pyrazinamide | 3.125 |
| Ciprofloxacin | 3.125 |
| Streptomycin | 6.25 |
*Minimal Inhibitory Concentration
Table 1: In vitro anti-tubercular activity of the synthesized compounds (7a-p) against Mycobacterium tuberculosis (H37RV strain)
It was found that compounds substituted with aromatic/cyclic derivatives (like cyclopropane, phenyl, morpholine) showed potent activity than aliphatic i.e., acetate, sulfonyl, alkyl substituents. Based on structural similarities with the first line anti-tuberculosis drugs (i.e., pyrazinamide), the synthesized compounds may act by inhibiting ATP synthesis and also acidifies M. tuberculosis to inhibit translation [17].
Conclusion
In this study, novel indolizine derivatives were designed, synthesized and screened for in vitro antimycobacterial activity against M. tuberculosis H37Rv strain. The structures of the synthesized compounds were characterized by 1H-NMR and Mass spectroscopic results. All compounds screened for anti-TB activity using MABA, the compounds 7a-p showed anti-tubercular activity within MIC range of 1.6 to 6.25 μg/ml, more or equivalent to those for standard anti-Tb drugs (pyrazinamide, ciprofloxacin with MICs 3.125 μg/ml; streptomycin MIC 6.25 μg/ml). From the above results, due to the presence of novel structure and potent anti-tubercular activity it can be concluded that the tested compounds could be a best initiation to find new lead to class of anti-tubercular agents in the future.
Acknowledgement
The authors acknowledge Maratha Mandal Dental College, Belgaum for biological screening support.
References
- S.H.E. Kaufmann, Nature, 2008, 453, 295-296.
- J. Mao, H. Yuan, Y. Wang, B. Wan, D. Pak, R. He, S.G. Franzblau, Bioorganic & Medicinal Chemistry Letters, 2010, 20, 3, 1263-1268.
- T. Beghyn, R. Deprez-Poulain, N. Willand, B. Folleas, B. Deprez, Chem. Biol. Drug Des., 2008, 72, 1, 3-15.
- S. Muthusaravanan, S. Perumal, P. Yogeeswari, D. Sriram, Tetrahedron Lett., 2010, 51, 49, 6439-6443.
- Y.M. Shen, P.C. Lv, W. Chen, P.G. Liu, M.Z. Zhang, H.L. Zhu, Eur. J. Med. Chem., 2010, 45, 7, 3184-3190.
- P. Sonnet, P. Dallemagne, J. Guillon, C. Enguehard, S. Stiebing, J. Tanguy, R. Bureau, S. Rault, P. Auvray, S. Moslemi, P. Sourdaine, G.E. Séralini, Biorg. Med. Chem., 2000, 8, 5, 945-955.
- S.C. Smith, E.D. Clarke, S.M. Ridley, D. Bartlett, D.T. Greenhow, H. Glithro, A.Y. Klong, G. Mitchell, G.W. Mullier, Pest Management Science, 2005, 61, 1, 16-24.
- Antonini, M. Cardellini, F. Claudi, P. Franchetti, U. Gulini, G. DeCaro, F. Venturi, J. Pharm. Sci., 1977, 66, 12, 1692-1696.
- K.M. Dawood, H. Abdel-Gawad, M. Ellithey, H.A. Mohamed, B. Hegazi, Arch. Pharm. Pharm. Med. Chem., 2006, 339, 3, 133-140.
- S. Chen, Z. Xia, M. Nagai, R. Lu, E. Kostik, T. Przewloka, M. Song, D. Chimmanamada, D. James, S. Zhang, J. Jiang, M. Ono, K. Koya, L. Sun, Med. Chem. Commun., 2011, 2, 176-180.
- M.C. Wani, A.W. Nicholas, M.E. Wall, J. Med Chem., 1987, 30, 12, 2317-2319.
- M.M. Badran, K.A.M. Abouzid, M.H.M.Hussein, Arch. Pharm. Res., 2003, 26, 107-113.
- M.N.A.Nasr, Arch. Pharm. Pharm. Med.Chem., 2002, 335, 8, 389-394.
- S.A. El-Hawash, N.S. Habib, N.H. Fanaki, Die Pharmazie, 1999, 54, 11, 808-813.
- A.A. El-Gendy, S. El-Meligie, A.K. El-Ansary, A.M. Ahmedy, Arch. Pharm. Res., 1995, 18, 1, 44-47.
- M.C.S. Lourenco, M.V.N. DeSouza, A.C. Pinheiro, M.L. Ferreira, R.S.B. Goncalves, T.C.M. Nogueira, M. A. Peralta, Arkivoc, 2007, xv, 181-191.
- N. Arya, M.K. Raut, S.G. Tekale, C.J. Shishoo, K.S. Jain. Austin J. Anal. Pharm. Chem., 2014, 1, 3, 1014.
Supplementary Data
In vitro Anti-tubercular activity