Research Article - Der Pharma Chemica ( 2019) Volume 11, Issue 6
Synthesis and Spectral Studies of Some Novel 9-Benzylpurine Derivatives
Arvinder Pal Singh* and Noolvi MNArvinder Pal Singh, Department of Pharmaceutical Chemistry, Rayat Institute of Pharmacy, Railmajra. S.B.S Nagar, Punjab-144 533, India, Tel: 9417667810, Email: arvsodhi19@gmail.com
Received: 22-Apr-2020 Accepted Date: Apr 30, 2020 ; Published: 13-Nov-2019
Abstract
Preparation of 9-benzylpurine derivatives is a tough task to any synthetic chemist. 2, 6-dichloropurine is firstly prepared for synthesizing novel 9-benzylpurine derivatives. This dihalopurine can be prepared from both Xanthine and Adenine and their comparison w.r.t percentage yield, purity was done. This prepared dihalopurine was benzylated and two isomers having N-Benzylation at 9- and 7-position of dihalopurine was reported. These formed two isomers were separated by column chromatography. After separation, we have synthesized 6-(2-furyl)-9benzylpurine by Stile Coupling reaction. Then, 6-(2-furyl)-9-benzylpurine was aminated to form novel 9-benzylpurine derivatives. The synthesized novel compounds were identified initially by TLC by marking spots of intermediate compound and final formed product on TLC plate using solvent CHCl3: Me OH (8.5:1.5). Then, their further characterization was done by I.R, 1HNMR, 13CNMR and Mass Spectral data.
Keywords
9-Benzylpurine, Anti-microbial, Chromatography, Stile coupling reaction, Spectral studies.
Introduction
Due to increasing anti-microbial resistance, such as to methicillin and vancomycin resistant to Staphylococcus aureus, vancomycin to enterococci, multi-drug target resistance to genus pseudomonas, bacteria belonging to klebsiella family, enterobacter (motile aerobic gram negative bacilli) species and acinetobacter species that include gamma proteobacteria as well as Streptococcus pneumonia having resistance to penicillin and other anti-bacterial, more and more efficient anti-microbial agents are required. This need leads to development of some novel anti-microbial agents. These anti-microbial1 agents are synthesized to retard the growth or killing of infectious organism with minimal side effect on the recipient. This infectious microbe produces many life threatening diseases like tuberculosis, leprosy, malaria, amoebiasis, giardiasis, trichomoniasis, heminthiasis, filiariasis etc. and other fungal and viral infections that affect our defense mechanism [1].
So, 9-benzylpurine was chosen as lead compound, as it has very broad spectrum of anti-microbial activity. It is reported in the past that their derivatives such as 2,8 disubstituted 6-(2-furyl)-9-(p-methoxybenzyl) purines have anti-mycobacterial2 activity, 9-(2-chloro-6-fluorobenzyl)-6- (substituted amino) purines have anti-coccidial3 activity and anti-convulsant4 activity, Trisubstituted purines as selective CDK1 inhibitors5, several modified 9-benzylpurines having carbon substituents at the 2-, 6- or 8-positions gives anti-viral, anti-cancer, anti-hypertensive effect or cytokines activities [2-10].
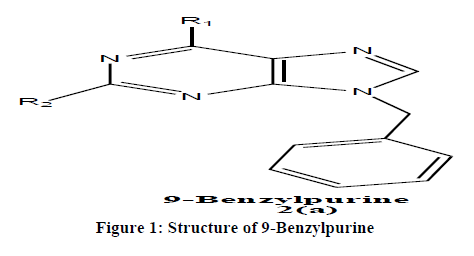
Chemically, 9-benzylpurines, 2 (a) is purine derivative, which consists of pyrimidine ring fused with imidazole ring with phenyl methyl substituents at 9-position (Figure 1).
Materials and Methods
The 1HNMR spectra were recorded at Sophisticated Analytical Instrumental Facility (SAIF) center, Punjab University, Chandigarh at 400 MHz, while 13CNMR were recorded at Central Instrumental Lab, N.I.P.E.R, Mohali at 100 MHz using Bruker Avance II 400 MHz NMR spectrometer. Both spectra have chemical shifts (δ) in ppm downfield w.r.t TMS. The solvent used for carrying out the 1HNMR and 13CNMR is CDCl3. Buchi rotavapour was used for the evaporation of solvents under vacuum. Melting point of the all prepared compounds has been determined by Perfit Melting Point apparatus. The borosil 6101065 chromatography column is used for separation of purification and separation of prepared compounds using Silica gel 40 grade. For Thin Layer Chromatography, TLC silica gel 60G F254 plates of Merck are used (Figure 2).
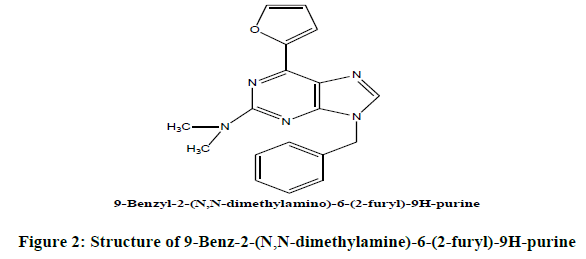
Target compound to be synthesized 5 (a)
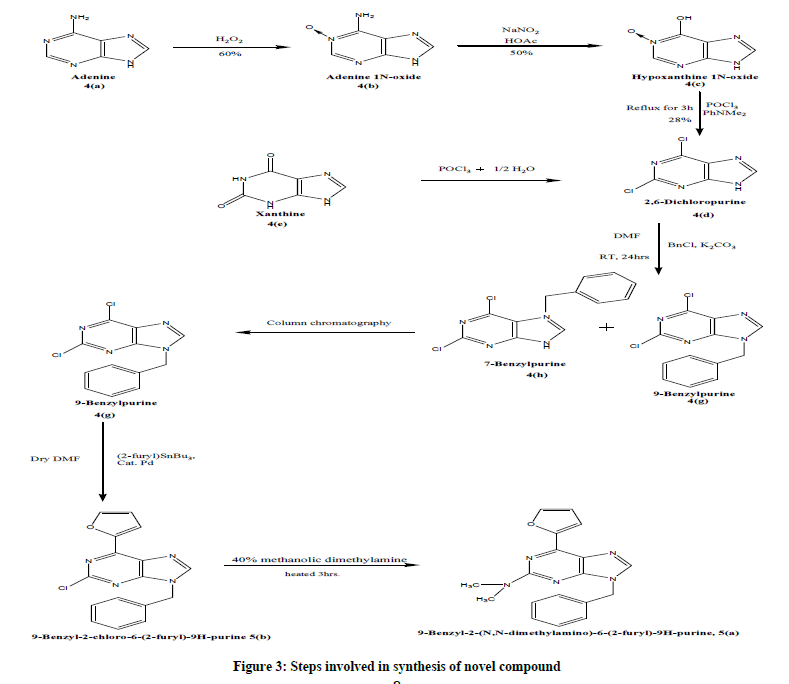
B) Steps involved in synthesis of novel compound (Figure 3).
Procedure for synthesizing target compound
2,6-dihalopurines synthesis (4d): 2, 6-dihalopurine was prepared by two starting materials xanthine and adenine [11-16].
Preparation of 2, 6-dichloropurines from xanthine: Firstly, 8 gm of xanthine is taken along with 64 ml of pyrophosphoryl chloride in a reflux condenser fitted RBF and heating is done at 165°C for 19 hrs. The resultant mixture was cooled down followed by the separation of brown solution from solid residue at the bottom. Then crushed ice is added to solid residue. The formed precipitates were removed and product was extracted out from filtrate using ether. The solvent was allowed to evaporate leaving behind solid product which was kept in desiccator for complete removal of moisture and solvent. A light yellow colored product obtained [6].
Preparation of pyrophosphoryl chloride: Added 20 ml of water to 200 ml of phosphoryl chloride drop-wise. This reaction mixture was boiled for 1.5 hrs to expel out all formed HCl gas. The reaction mixture was allowed to cool. After cooling, top layer was separated from bottom layer. A thick syrupy viscous solution remains at the bottom, which is used for xanthine chlorination [6].
Preparation of 2,6-dichloropurines from commercial available Adenine
Preparation of Adenine 1-N-oxide: While maintaining temperature below 200 C, 10 gm of adenine was dissolved in 60 ml of hot acetic acid. Then, 30% of hydrogen peroxide (37 ml approx.) was added slowly. This solution was kept at room temperature for 2.5 days. Crystals were formed, which are collected and washed with little acetic acid. Recrystallization of the material was done with boiling water and adenine-1Noxide (5 gm) was formed [7].
Preparation of hypoxanthine 1N-oxide: Above prepared, Adenine 1N-oxide was dissolved in a hot aqueous glacial acetic acid (100 ml). An aq. solution of sodium nitrite was added to the above mixture by maintain its temperature below 200 C. After the addition of sodium nitrite, it was kept at room temperature for 4 days. Pale yellow crystals were formed, which were filtered, washed with hot water and dried [8].
Preparation of 2, 6-dichloropurine: A mixture of hypoxanthine 1N-oxide (1.034 gm), triethylamine (1.5 gm) and phosphoryl chloride (60 ml) was refluxed for 3 hrs. The formed pale brown residue was poured into ice water and the volume was makeup up to 100 ml. This aqueous mixture was made strongly alkaline with 10 N NaOH solution. The liberated triethylamine was extracted out with ether and then, aq. layer was made slightly acidic with conc. HCl. This acidic reaction mixture was taken in separating funnel for extraction with ether. The ethereal layer was eluted, collected, dried. A yellow powder was obtained after evaporation of ether [9].
Synthesis of 9-benzyl-2, 6-dichloropurines10 (4 g): Dihalopurine (1.528 gm) was dissolved in 50 ml of dried DMF and potassium carbonate (3.40 gm) was added slowly while stirring at room temperature in the presence of nitrogen. After 20 min, benzyl chloride (1.26 ml) was added and then, 40 hrs. of continuous stirring was done. After that, it was filtered and solvent was evaporated under vacuum. This formed product contained two isomers i.e. 9-benzylpurine and 7-benzylpurine, which was separated by column chromatography using mobile phase Ethyl acetate-acetone-hexane (1.5:3:10).
Synthesis of 2-chloro-9-Benzyl-6-(furan-2-yl)-9H-purine10 (5b): Added 9-benzyl-2,6-dichloro-9H-purine (279 mg), bis (triphenylphosphine)pd (II)chloride (35 mg) and tributylstannylfuran (0.17 ml) in a conical flask fitted with reflux condenser and heated at 50°C under N2 for 3 days with solvent dried DMF (3 ml). Then, a saturated methanolic solution of KF (20 ml) was added drop wise and stirred at room temperature overnight. After evaporation of solvent, light yellow coloured crystals formed. Purification of this formed crystals were done using column chromatography with mobile phase ethyl acetate-hexane (4:10).
Synthesis of 9-Benzyl-2-(N, N-dimethyl amino)-6-(furan-2-yl)-9H-purine11 (5a): A mixture of 9-benzyl-2-chloro-6 (furan-2-yl)-9H-purine (50 mg), 40% methanolic dimethyl amine (186 mg) and Me OH (5 ml) was heated for 3 h at 110-120°C. Then, cooled and the solution was allowed to evaporate in vacuum. The crystalline residue was obtained. The synthesized dimethyl amino novel compounds were identified initially by TLC by marking spots of intermediate compound and final formed product on TLC plate using solvent CHCl3: Me OH (8.5:1.5) [14- 20].
Conclusion
Synthesis of dichloropurine from xanthine gives high percentage yield than via adenine. Moreover, dichloropurine obtained from xanthine is purer than from adenine. Synthesized novel compound i.e. 9-Benzyl-6-(furan-2-yl)-2 (N, N-dimethyl amino)-9H-purine had dimethyl amine substituent at 2-position. This dimethyl amine moiety along with purine derivative inhibits the growth of coccidiosis to some extent and increasing this alkyl chain decreases the activity [11]. More future studies on 9-benzylpurine derivatives as anti-microbial agents would prove beneficial.
Acknowledgment
The authors are grateful to the Department of Pharmacy, Rayat Institute of Pharmacy, Railmajra, S.B.S Nagar for providing the research facility for this investigation.
References
[1] K.D. Tripathi: Essentials of Medical Pharmacology, Jaypee brothers medical publishers, Delhi, 5th edition, 2005, 771-824
[2] M. Braendvang and Gunderson Lise-Lotte, Bioorg. Med. Chem., 2007, 15, 7144
[3] Imai Kin-ichi and Seo Takuji, Eur.J.Med.Chem.-Chimica Therapeutic, 1980, 15: 207
[4] J.L. Kelly, M.P. Krochmal, J.A. Linn et al., J.Med.Chem. 1988,31:606
[5] P. Imbach, H.G. Carpraro, P. Furet et al., J.Bioorg.Med.Chem.Lett. 1999, 9, 91-96
[6] N.K. Saxena, P.K. Gupta and D.S Bhakuni, Indian J.Chem. 1980, 19B, 332
[7] M.A. Stevens, D.I. Magrath, H.W. Smith et al., J. Am.Chem.Soc., 1958, 80, 2755
[8] H. Kawashima, T. Meguro and I. Kumashiro, United States Patent Office, 3317511, 1967
[9] H. Kawashima and I. Kumashiro, United States Patent Office, 3314938, 1967.
[10] Langli Geir, Gunderson Lise-Lotte and Rise Forde, Tetrahedron, 1996, 52 (15), 5625
[11] Imai Kin-ichi and Seo Takuji, Eur.J.Med.Chem.-Chimica Therapeutic. 1980, 15 (3), 207
[12] G.B. Elion and Mitclungs, J. Am.Chem.Soc. 1956, 78: 3508
[13] Guo Hai-Ming, Pu Li, Niu Hong-Ying et al., The Journal of Organic Chemistry. 2010, 75 (17), 6016
[14] Leire Aguado, Hendrik Jan Thibaut, Eva-Maria Priego et al., Journal of Medicinal Chemistry. 2010, 53 (1), 316
[15] Zhong-Zhen Zhou, Xiu-Dong Shi, Hong-Fang Feng et al., European Journal of Medicinal Chemistry. 2017, 138, 1126
[16] Anders Foller Larsen and Trond Ulven, Chemical Communications. 2014, 50, 4997
[17] J. Dodonova, I. Uogintaite, V. Masevicius et al., Chemistry of Heterocyclic Compounds. 2010, 46, 1122
[18] M.S. Prem Chauhan, Naresh Sunduru and Moni Sharma, Future Medicinal Chemistry. 2010, 2 (9), 1469
[19] Vitezslav Bambuch, Radek Pohl and Michal Hocek, European Journal of Organic Chemistry. 2008, 2783
[20] Morten Braendvang and Lise-Lotte Gundersen, Bioorganic & Medicinal Chemistry. 2007, 15, 7144
[21] L. Yves Janin. Antituberculosis drugs, Ten years of research. Bioorganic & Medicinal Chemistry. 2007, 15, 2479
[22] Morten Brændvang and Lise-Lotte Gundersen, Bioorganic & Medicinal Chemistry. 2005, 13, 6360.
[23] M. Morivasu, K. Kawanishi, A. Kato et al., Chem. SOC. Japan. 1984, 57, 1766
[24] J. L. Kelley, M. P. Krochmal, J. A. Linn et al., J. Med. Chem. 1988, 31, 606
[25] W. C. Still, M. Kahn and A. J. Mitra, Org. Chem. 1978, 43, 2923



