Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 3
Synthesis and Utility of Naphthalen-benzofuran Chalcone in the Synthesis of New Pyrazole, Isooxazole, Thiazole, Pyrimidine, Pyran, Pyridine and Different Azide Derivatives with Antiviral and Antitumor Activity
Rasha S Gouhar1*, Ewies F Ewies2, Mohamed F El-Shehry3, El-Mahdy M El-Mahdy4 and Mohamed NF Shaheen4
1Therapeutic Chemistry Department, National Research Centre, 12622, Dokki, Giza, Egypt
2Department of Organometallic and Organometalloid Chemistry, National Research Centre, 12622, Dokki, Giza, Egypt
3Pesticide Chemistry Department, National Research Centre, 12622, Dokki, Giza, Egypt
4Environmental Virology Laboratory, Water Pollution Research Department, National Research Centre, 12622, Dokki, Giza, Egypt
- *Corresponding Author:
- Rasha S Gouhar
Therapeutic Chemistry Department
National Research Centre
12622, Dokki, Giza, Egypt
Abstract
Synthesis and utility of naphthalene-benzofuran chalcone in different types of reactions for syntheses of various heterocyclic compounds such as pyrazole, isooxazole, thiazole, pyrimidine, pyran, pyridine rings were done. Also, formation of azide derivatives from pyridine acetohydrazide that used in synthesis of 2,4-dioxothiazolidine and 2,4-dioxo-1,2-dihydroquinazolin derivatives were run and reacted with different types of phenol and amine such as phenol, 4-aminophenol, p-chloroaniline and 4-aminopyridine respectively. The structures of the newly synthesized compounds were confirmed based on different spectroscopic and elemental analyses (IR, 1H, 13C-NMR and MS). Selected new compounds were evaluated their activity as antiviral against Hepatitis A Virus (HAV) using 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) and Tissue Culture Infectious Doses (TCID50) assays. These compounds showed high to moderate activity of antiviral activity. In addition, The antitumor activities of certain selected new compounds were screened, in vitro, against a panel of human solid tumor cell lines (U937: Cell line human lymphoblast lung from human, MOLT-4: Human lymphoblast; Acute lymphoblastic leukemia, K562: Cell line human leukemic blood from human).
Keywords
Chalcone, Pyrazole, Isooxazole, Thiazole, Pyrimidine, Pyran, Pyridine, Azide derivatives, HAV antiviral, Antitumor activity
Introduction
Chalcone is an α, β-unsaturated carbonyl system, that is a bi-electrophilic and keto-vinyl chain between two rings and highly reactive and acts as an vital chemical synthon for synthesis of different five, six and seven membered heterocyclic compounds that containing different heteroatoms such as nitrogen, oxygen and sulfur atoms by abridgment with a variety of bi-nucleophilic reagents [1-3]. The compounds with chalcone as backbone have been cited to possess varied biological and pharmacological activities, including antimicrobial, anti-inflammatory, analgesic, cytotoxic, antitumor, antimalarial, antitubercular, antiviral, anti-HIV, antiulcerative, antileishmanial, antioxidant, antiprotozoal, antihistaminic, antifedent, immunomodulatory, anticonvulsant, antihyperglycemic, antihyperlipidemic and antiplatelet activities [3-7]. Thus, chalcones continue to attract considerable scientific attention because of their association with a variety of biological activities.
On the other hand, molecular hybridization is a vital strategy, which has been successfully applied for development and synthesis of efficient scaffolds that are chemotherapeutic agents [8]. This strategy involves the link of two or more chemical entities to form new hybrid moieties [9]. The new designed chemical moieties were selected on the basis of their expected or known bio-profiles with its additive pharmacological activities [10,11].
Besides that, Hepatitis A virus (HAV) infects the liver, belongs to the genus hepatovirus within the picornaviridae family and it is a single RNA strand covered by icosahedral shaped protein shell [12]. The search of new synthetic compounds to overcome HAV is continues during the last decades [13-15].
Also, naphthalene derivatives have attracted significant attention in the field of medicinal chemistry due to their wide applications in drug discovery [16-21]. Pyrazole, thiazole, isooxazole and pyridine or pyrimidine derivatives have been the subject of medical research due to their various biological and pharmacological properties [22-27].
Based on the survey, we report herein the synthesis of some new naphthalene-benzofuran derivatives bearing various heterocyclic compounds such as pyrazole, isooxazole, thiazole, pyrimidine, pyran, pyridine moieties and some of the newly synthesized derivatives were tested as antiviral agents against Hepatitis A Virus (HAV). In addition, the cytotoxic activity of some prepared derivatives against U937, Molt 4 and K562 cancer cell lines was evaluated.
Experimental Section
All melting points are uncorrected and were taken in open capillary tubes using Electrothermal apparatus 9100. Elemental microanalyses were carried out at Microanalytical Unit, Central Services Laboratory, National Research Centre, Dokki, Giza, Egypt, using Vario Elementar and were found within ± 0.4% of the theoretical values. Infrared spectra were recorded on a Jasco FT/IR-6100, Fourier transform, The IR spectra were measured in KBr pellets with a Perkin-Elmer Infracord Spectrophotometer model 157 (Grating). Routine NMR, 1H-NMR (500 MHz) and 13C, 1H-NMR (125 MHz) spectra were recorded at room temperature on a JEOL-500 MHz Spectrometer as solutions in Deuterated chloroform (CDCl3)/or Dimethyl Sulfoxide (DMSO). All chemical shifts are quoted in δ relative to the trace resonance of protonated chloroform (CDCl3), (δ=7.25 ppm) and (δ=77.0 ppm), protonated DMSO (δ=2.50 ppm) and (δ=39.51 ppm). The mass spectra were measured with a GC Finnigan MAT SSQ-7000 mass spectrometer. Follow up of the reactions and checking the purity of the compounds were made by TLC on silica gel-precoated aluminum sheets (Type 60F254, Merck, Darmstadt, Germany) and the spots were detected by exposure to UV lamp at λ254 nanometer for few seconds. The chemical names given for the prepared compounds are according to the IUPAC system. Solvents were dried/purified according to conventional procedures. Some of the experimental part was obtained according to published methods [28,29].
Synthesis of 1-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)ethanone (1)
To a solution of salicyaldehyde (2.94 g; 1 mmol) in acetone (40 ml), chloroacetone (0.93 g; 1 mmol) and K2CO3 (2.0 g) were added. The solution was heated under reflux temperature and suspended in ice-cooled water. The solid product obtained was collected by filtration and crystallized from ethanol to give the product (1).
Yield: 74%; M.p. 79-81°C. IR (KBr): Ṽ =1689 (C=O), 1591 (N=N), 1270 (C-F) cm-1; 1H-NMR (500 MHz, DMSO), δ=8.18-6.68 (m, 8H, Harom), 2.52 (s, 3H, CH3); 13C-NMR (125 MHz, DMSO), δ=188.4 (C=O), 158.4 (cyclic C-O), 155.2 (cyclic C-O), 152.8 (cyclic C-N), 146.6 (cyclic C-N), 131.2, 130.2, 129.7, 128.2, 127.8, 127.0, 126.4, 118.8, 114.8, 112.9 (Ar-C), 26.8 (CH3). MS (EI, 70 eV): m/z (%)=332 (5%) [M]+. Anal. Calcd. For C17H11F3N2O2 (332.28): C, 61.45; H, 3.34; N, 8.43; Found: C, 61.54; H, 3.48; N, 8.54.
Synthesis of naphthalen-benzofuran chalcone 3
A mixture of acetyl benzofuran 1 (0.01 mol) and naphthyl-4-carboxaldehyde 2 (0.01 mol) in alcoholic sodium hydroxide (10%, 200 ml) was stirred overnight at room temperature. The formed precipitate was filtered, washed several times with water, dried and recrystallized from ethanol to give the title compound 3.
3-(Naphthalen-2-yl)-1-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)prop-2-en-1-one (3): Product 3 was separated as red
crystals, yield 75%. M.p. 182-183°C. IR (KBr): Ṽ = 1655 (C=O), 1640 (C=C), 1590 (N=N), 1275 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.17-7.38 (m, 15H, Harom), 7.74 (d, 1H, CH=), 6.42 (d, 1H, =CH); 13C-NMR (125 MHz, CDCl3), δ=178.4 (C=O), 161.4 (cyclic C-O), 156.2 (cyclic C-O), 151.8 (cyclic C-N), 147.6 (cyclic C-N), 144.5, 134.9, 133.8, 132.4, 131.3, 130.2, 129.7, 128.8, 127.1, 126.4, 124.2, 119.8, 117.8, 115.8, 112.9 (Ar-C). MS (EI, 70 eV): m/z(%)=470 (5%) [M]+, 289 (100%) [M-181]+. Anal. Calcd. For C28H17F3N2O2 (470.44): C, 71.49; H, 3.64; N, 5.95; Found: C, 71.50; H, 3.61; N, 5.92.
5-(Naphthalen-2-yl)-3-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)-4,5-dihydro-1H-pyrazole (4)
A mixture of compound 3 (0.001 mol) and hydrazine hydrate 98% (0.002 mol) in absolute ethanol (10 ml) was refluxed for 4 hr. The formed precipitate was filtered, dried and recrystallized from ethanol to give the title compound 4.
Product 4 was separated as yellow crystals, yield 65%. M.p. 133-134°C. IR (KBr): Ṽ =3350 (NH), 1635 (C=C), 1560 (N=N), 1270 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3) δ=10.38 (s, 1H, NH, exchangeable with D2O), 8.27-7.21 (m, 16H, Harom), 5.12 (dd, 1H, CH-pyrazoline proton), 3.10, 3.35 (2dd, 2H, CH2-pyrazoline protons); 13C-NMR (125 MHz, CDCl3), δ=161.2 (cyclic C=N), 146.1 (cyclic C-O), 142.6, 134.5, 134.1, 132.5, 131.3, 130.7, 129.7, 127.9, 127.2, 124.1, 122.7, 121.7, 119.7, 117.2, 115.1, 112.1 (Ar-C), 106.8 (CH, furan), 67.2 (CH, pyrazoline), 40.1 (CH2 pyrazoline). MS (EI, 70 eV): m/z (%)=484 (15%) [M]+, 303 (100%) [M-181]+. Anal. Calcd. For C28H19F3N4O (484.47): C, 69.42; H, 3.95; N, 11.56; Found: C, 69.45; H, 3.99; N, 11.52.
1-(5-(Naphthalen-2-yl)-3-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)-4,5-dihydro-1H-pyrazol-1-yl)ethanone (5)
A mixture of compound 3 (0.001 mol) and hydrazine hydrate 98% (0.002 mol) in glacial acetic acid (10 ml) was refluxed for 4 h. The formed precipitate was filtered, dried and recrystallized from acetic acid to give the title compound 5.
Product 5 was separated as yellow crystals, yield 65%. M.p. 145-146°C. IR (KBr): Ṽ =1722 (C=O), 1620 (C=C), 1560 (N=N), 1260 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.31-7.39 (m, 16H, Harom), 5.10 (dd, 1H, CH-pyrazoline proton), 3.15, 3.30 (2dd, 2H, CH2-pyrazoline protons), 2.28 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3); δ=163.70 (C=O), 151.8 (cyclic C-N), 146.2 (cyclic C-O), 147.6 (cyclic C-N), 143.9, 135.2, 134.0, 131.0, 129.7, 128.2, 127.0, 126.5, 124.1, 121.1, 119.8, 117.8, 115.7, 113.8 (Ar-C), 112.6 (CH, furan), 67.5 (CH, pyrazoline), 40.6 (CH2 pyrazoline), 23.5 (CH3). MS (EI, 70 eV): m/z(%)=526 (35%) [M]+. Anal. Calcd. For C30H21F3N4O2 (526.51): C, 68.44; H, 4.02; N, 10.64; Found: C, 68.48; H, 4.00; N, 10.69.
General procedures for synthesis of 6a-c
A mixture of compound 3 (0.001 mol) and the appropriate hydrazine namely; methyl hydrazine, phenyl hydrazine and/or 2,4-dinitrophenyl hydrazine (0.002 mol) in absolute ethanol (10 ml) was refluxed for 3 h. After cooling, the formed precipitate was filtered, dried and recrystallized from ethanol to give the title compounds 6a-c.
1-Methyl-5-(naphthalen-2-yl)-3-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)-4,5-dihydro-1H-pyrazole (6a): Product 6a was separated as reddish brown crystals, yield 66%. M.p. 152-153°C. IR (KBr): Ṽ =1625 (C=C), 1565 (N=N), 1260 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.21-7.37 (m, 15H, Harom), 3.72 (dd, 1H, CH), 2.90 (dd, 1H, CH2), 2.66 (dd, 1H, CH2), 2.51 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3); δ=156.6 (cyclic C-O), 152.7 (C=N, pyrazole), 151.4 (cyclic C-N), 147.6 (cyclic C-N), 143.4 (cyclic C-O), 136.7, 134.9, 133.3, 131.7, 129.3, 128.7, 127.0, 126.3, 124.8, 122.6, 121.0, 119.3, 117.1, 115.7, 112.6 (Ar-C), 102.5 (CH, furan), 70.2 (CH, pyrazole), 40.4 (CH2, pyrazole), 39.0 (CH3). MS (EI, 70 eV): m/z (%)=498 (15%) [M]+, 209 (100%) [M-289]+. Anal. Calcd. For C29H21F3N4O (498.50): C, 69.87; H, 4.25; N, 11.24; Found: C, 69.90; H, 4.28; N, 11.20.
5-(Naphthalen-2-yl)-1-phenyl-3-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)-4,5-dihydro-1H-pyrazole (6b): Product 6b was separated as yellow crystals, yield 60%. M.p. 109-111°C. IR (KBr): Ṽ =1615 (C=C), 1568 (N=N), 1250 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3); δ=8.20-7.06 (m, 19H, Harom), 6.21 (s, 1H, CH, furan), 4.73 (t, 1H, CH), 3.02 (dd, 1H, CH2), 2.80 (dd, 1H, CH2); 13C-NMR (125 MHz, CDCl3), δ=159.9 (C=N, pyrazole), 156.6 (cyclic C-O), 151.4 (cyclic C-N), 147.1 (cyclic C-N), 143.1 (cyclic C-O), 142.0, 138.1, 134.9, 133.3, 131.2, 129.7, 128.7, 127.0, 126.3, 124.4, 122.6, 121.1, 119.3, 117.1, 117.6, 115.7, 112.6 (Ar-C), 102.5 (CH, furan), 70.5 (CH, pyrazole), 40.4 (CH2, pyrazole). MS (EI, 70 eV): m/z (%)=560 (35%) [M]+, 271 (100%) [M-289]+. Anal. Calcd. For C34H23F3N4O (560.57): C, 72.85; H, 4.14; N, 9.99; Found: C, 72.81; H, 4.17; N, 9.95.
1-(2,4-Dinitrophenyl)-5-(naphthalen-2-yl)-3-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)-4,5-dihydro-1H-pyrazole (6c): Product 6c was separated as reddish orange crystals, yield 72%. M.p. 171-172°C. IR (KBr): Ṽ =1630 (C=C), 1555 (N=N), 1370 (NO2), 1255 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.97-7.35 (m, 17H, Harom), 6.11 (s, 1H, CH, furan), 4.72 (t, 1H, CH), 3.03 (dd, 1H, CH2), 2.81 (dd, 1H, CH2); 13C-NMR (125 MHz, CDCl3), δ=159.77 (C=N, pyrazole), 156.4 (cyclic C-O), 151.4 (cyclic C-N), 147.1 (cyclic C-N), 143.1 (cyclic C-O), 138.0, 134.9, 133.8, 131.2, 129.7, 128.4, 127.0, 126.9, 125.5, 123.1, 119.3, 117.3, 115.7, 112.9 (Ar-C), 102.5 (CH, furan), 70.6 (CH, pyrazole), 41.1 (CH2, pyrazole). MS (EI, 70 eV): m/z (%)=650 (45%) [M]+. Anal. Calcd. For C34H21F3N6O5 (650.56): C, 62.77; H, 3.25; N, 12.92; Found: C, 62.80; H, 3.21; N, 12.35.
5-(naphthalen-2-yl)-3-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)isoxazole (7)
A mixture of compound 3 (0.002 mol) and hydroxylamine hydrochloride (0.002 mol) in alcoholic sodium hydroxide (5%, 20 ml) was refluxed for 10 h. The reaction mixture was cooled, poured onto ice/cold water and acidified by diluted hydrochloric acid. The formed precipitate was filtered, dried and recrystallized from ethanol to give the title compound 7.
Product 7 was separated as yellow crystals, yield 75%. M.p. 130-132°C. IR (KBr): Ṽ =1660 (C-O), 1618 (C=C), 1580 (C=N), 1550 (N=N), 1255 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.30-7.39 (m, 16H, Harom); 13C-NMR (125 MHz, CDCl3), δ=172.1 (C-O, isoxazole), 156.1 (C-O, furan), 154.8 (C=N, isoxazole), 151.8 (C-N), 149.3 (C-O, furan), 147.6 (C-N), 134.5, 134.0, 131.2, 130.4, 129.7, 128.7, 127.9, 126.5, 124.1, 122.7, 120.4, 119.8, 117.8, 116.5 (Ar-C), 112.7 (CH, furan), 103.0 (CH, isoxazole). MS (EI, 70 eV): m/z (%)=483 (65%) [M]+. Anal. Calcd. For C28H16F3N3O2 (483.44): C, 69.56; H, 3.34; N, 8.69; Found: C, 69.53; H, 3.33; N, 8.71.
General Procedures for synthesis of 8a, b
A mixture of compound 3 (0.4 g, 0.001 mol), semicarbazide hydrochloride (0.001 mol) and/or thiosemicarbazide (0.004 mol) and sodium acetate (0.001 mol, in case of 8a) in absolute ethanol (15 ml) was refluxed for 4-6 h. Then the reaction mixture was poured onto ice/cold water. The formed precipitate was filtered, dried and recrystallized from ethanol to give the title compound 8a, b.
5-(Naphthalen-2-yl)-3-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)-1H-pyrazole-1-carboxamide (8a): Product 8a was separated as yellow crystals, yield 70%. M.p. 145-146°C. IR (KBr): Ṽ =3533 (NH2), 1675 (C=O), 1615 (C=C), 1570 (C=N), 1530 (N=N), 1245 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.23-7.40 (m, 16H, Harom), 4.88 (s, 2H, NH2, exchangeable with D2O); 13C-NMR (125 MHz, CDCl3), δ=160.9 (C=N, pyrazole), 156.1 (C=O), 153.9 (cyclic C-O), 151.8 (cyclic C-N), 148.0 (cyclic C-O), 147.6 (cyclic C-N), 140.4, 135.2, 134.0, 131.7, 129.9, 128.2, 127.0, 124.4, 122.6, 121.2, 119.3, 117.1, 115.7 (Ar-C), 109.4 (CH, pyrazole), 107.5 (CH, furan). MS (EI, 70 eV): m/z(%)=525 (35%) [M]+. Anal. Calcd. For C29H18F3N5O2 (525.48): C, 66.28; H, 3.45; N, 13.33; Found: C, 66.30; H, 3.48; N, 13.30.
5-(Naphthalen-2-yl)-3-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)-1H-pyrazole-1-carbothioamide (8b): Product 8b was separated as yellow crystals, yield 72%. M.p. 165-166°C. IR (KBr): Ṽ =3531 (NH2), 1615 (C=C), 1570 (C=N), 1535 (N=N), 1271 (C=S), 1245 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.30-7.34 (m, 16H, Harom), 6.90 (s, 2H, NH2, exchangeable with D2O); 13C-NMR (125 MHz, CDCl3), δ=171.1 (C=S), 165.6 (C=N, pyrazole), 156.1 (C-O, furan), 151.8 (C-N=), 148.0 (C-O, furan), 147.6 (C-N=), 139.9, 135.2, 134.5, 131.2, 129.7, 128.2, 127.9, 126.5, 124.3, 122.1, 121.0, 117.1 (Ar-C), 112.9 (CH, pyrazole), 107.3 (CH, furan). MS (EI, 70 eV): m/z (%)=541 (75%) [M]+. Anal. Calcd. For C29H18F3N5OS (541.55): C, 64.32; H, 3.35; N, 12.93; Found: C, 64.30; H, 3.33; N, 12.90.
2-(5-(Naphthalen-2-yl)-3-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)-1H-pyrazol-1-yl)-4-phenylthiazole (9)
A mixture of compound 8b (0.001 mol), phencyl bromide (0.001 mol) in absolute ethanol (15 ml) was refluxed for 5 h. Then the reaction mixture was cooled, poured onto ice/cold water. The formed precipitate was filtered, washed several times by diluted ammonia solution, filtered, dried and recrystallized from ethanol to give the title compound 9.
Product 9 was separated as yellowish brown crystals, yield 68%. M.p. 181-182°C. IR (KBr): Ṽ =1613 (C=C), 1564 (C=N), 1535 (N=N), 1245 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.31-7.30 (m, 22H, Harom); 13C-NMR (125 MHz, CDCl3), δ=166.5 (C=N, pyrazole), 164.4 (C=N, thiazole), 156.1 (cyclic C-O), 151.8 (cyclic C-N), 148.0 (cyclic C-O), 147.6 (cyclic C-N), 144.1, 138.30, 135.2, 134.6, 131.2, 129.1, 128.22, 128.2, 127.9, 127.65, 126.5, 124.1, 124.1, 122.7, 121.1, 119.8, 117.8, 115.7, 112.9 (Ar-C), 111.2 (CH, thiazole), 107.4 (CH, furan), 104.4 (CH, pyrazole). MS (EI, 70 eV): m/z (%)=641 (35%) [M]+. Anal. Calcd. For C37H22F3N5OS (641.66): C, 69.26; H, 3.46; N, 10.91; Found: C, 69.30; H, 3.42; N, 10.93.
6-(Naphthalen-2-yl)-4-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)pyrimidine-2(1H)-thione (10)
A mixture of compound 3 (0.002 mol) and thiourea (0.002 mol) in sodium ethoxide solution (5%, 15 ml) was refluxed for 24 h. The reaction mixture was cooled, poured onto ice/cold water and acidified by diluted hydrochloric acid. The formed precipitate was filtered, dried and recrystallized from ethanol to give the title compound 10.
Product 10 was separated as reddish orange crystals, yield 60%. M.p. 110-112°C. IR (KBr): Ṽ =3444 (NH), 1635 (C=C), 1564 (C=N), 1535 (N=N), 1245 (C-F), 1210 (C=S) cm-1; 1H-NMR (500 MHz, CDCl3), δ=10.63 (s, 1H, NH, exchangeable with D2O), 8.89 (s, 1H, CH, pyrimidine), 8.27 (s, 1H, CH, furan), 8.20-7.39 (m, 14H, Harom); 13C-NMR (125 MHz, CDCl3), δ=183.2 (C=S), 157.2 (cyclic C-O), 156.8 (C-NH, pyrimidine), 156.2 (cyclic C-O), 153.6 (cyclic C-N), 151.8 (cyclic C-N), 147.6 (cyclic C-N), 134.5, 134.4, 134.0, 131.27, 130.5, 129.7, 128.2, 127.0, 126.5, 124.1, 122.1, 119.8, 117.2, 115.6, 112.3, 103.1 (Ar-C), 112.6 (CH, furan), 100.2 (CH, pyrimidine). MS (EI, 70 eV): m/z (%)=526 (75%) [M]+. Anal. Calcd. For C29H17F3N4OS (526.53): C, 66.15; H, 3.25; N, 10.64; Found: C, 66.19; H, 3.22; N, 10.61.
4-(Naphthalen-2-yl)-6-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)pyrimidin-2-amine (11)
A mixture of compound 3 (0.004 mol) and guanidine sulfate (0.004 mol) in sodium ethoxide solution (5%, 30 ml) was refluxed for 24 h. The reaction mixture was cooled, poured onto ice/cold water and acidified by diluted hydrochloric acid. The formed precipitate was filtered, dried and recrystallized from ethanol to give the title compound 11.
Product 11 was separated as yellow crystals, yield 72%. M.p. 187-188°C. IR (KBr): Ṽ =3554 (NH2), 1630 (C=C), 1560 (C=N), 1530 (N=N), 1240 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.17-7.37 (m, 14H, Harom), 6.97 (s, 1H, CH, pyrimidine), 6.55 (s, 1H, CH, furan), 1.63 (s, 2H, NH2, exchangeable with D2O); 13C-NMR (125 MHz, CDCl3), δ=162.5 (cyclic C-O), 161.4 (C-NH2), 159.6 (cyclic C-O), 156.1 (cyclic C-N), 152.0 (cyclic C-N), 134.5, 134.4, 134.0, 131.27, 130.5, 129.7, 128.2, 127.99, 127.0, 126.5, 124.14, 122.1, 119.8, 117.2, 115.6, 112.3, 103.1 (Ar- C), 102.39 (CH, pyrimidine). MS (EI, 70 eV): m/z (%)=509 (85%) [M]+. Anal. Calcd. For C29H18F3N5O (509.48): C, 68.37; H, 3.56; N, 13.75; Found: C, 68.40; H, 3.52; N, 13.72.
General procedures for synthesis of 12-13
A mixture of compound 3 (0.002 mol) and active methylene compounds namely; malononitrile, ethyl cyanoacetate and/or ethyl acetoacetate (0.002 mol) in sodium ethoxide solution (5%, 15 ml) was refluxed for 6 h. Then the reaction mixture was cooled, poured onto ice/cold water and acidified by diluted hydrochloric acid. The formed precipitate was filtered, dried and recrystallized from ethanol to give the title compounds 12- 13.
2-Amino-6-(naphthalen-2-yl)-4-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)-4H-pyran-3-carbonitrile (12): Product 12 was separated as reddish orange crystals, yield 63%. M.p. 120-122°C. IR (KBr): Ṽ =3571 (NH2), 2150 (CN), 1625 (C=C), 1570 (C-O), 1556 (N=N), 1245 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.95 (s, 2H, NH2, exchangeable with D2O), 8.20-7.39 (m, 14H, Harom), 6.65 (s, 1H, furan CH), 3.99 (d, 1H, pyran CH), 3.17 (d, 1H, pyran CH); 13C-NMR (125 MHz, CDCl3), δ=160.1 (C-O, furan), 154.31 (C-O, furan), 153.32 (C-O, pyran), 151.84 (C-N), 146.87 (C-O, pyran), 134.2, 131.2, 130.5, 129.7, 128.2, 127.9, 127.1, 127.0, 126.50, 126.2, 124.1, 122.5, 119.1, 117.0, 115.2, 112.5, 103.5 (Ar-C), 104.65 (CH, furan), 68.04 (C-CN). MS (EI, 70 eV): m/z (%)=536 (60%) [M]+. Anal. Calcd. For C31H19F3N4O2 (536.50): C, 69.40; H, 3.57; N, 10.44; Found: C, 69.42; H, 3.60; N, 10.41.
2-Hydroxy-6-(naphthalen-2-yl)-4-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)-4H-pyran-3-carbonitrile (13): Product 13 was separated as reddish orange crystals, yield 78%. M.p. 192-193°C. IR (KBr): Ṽ =3210 (OH), 2155 (CN), 1625 (C=C), 1640 (C-O), 1550 (N=N), 1233 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.15-7.25 (m, 14H, Harom), 6.50 (s, 1H, furan CH), 3.89 (d, 1H, pyran CH), 3.15 (d, 1H, pyran CH). 2.28 (s, 1H, OH, exchangeable with D2O); 13C-NMR (125 MHz, CDCl3), δ=160.1 (C-O, furan), 154.31 (C-O, furan), 153.32 (C-O, pyran), 151.84 (C-N), 146.87 (C-O, pyran), 134.2, 131.2, 130.5, 129.7, 128.1, 127.9, 127.2, 127.00, 126.5, 126.2, 124.1, 122.5, 119.8, 117.8, 115.8, 112.7, 103.9 (Ar-C), 104.5 (CH, furan), 68.1 (C-CN). MS (EI, 70 eV): m/z (%)=537 (60%) [M]+. Anal. Calcd. For C31H18F3N3O3 (537.49): C, 69.27; H, 3.38; N, 7.82; Found: C, 69.30; H, 3.40; N, 7.80.
1-(2-Hydroxy-6-(naphthalen-2-yl)-4-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)-4H-pyran-3-yl)ethanone (14): Product 14 was separated as yellow crystals, yield 80%. M.p. 207-209°C. IR (KBr): Ṽ =3215 (OH), 1710 (C=O), 1615 (C=C), 1650 (C-O), 1540 (N=N), 1230 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.20-7.26 (m, 14H, Harom), 6.80 (s, 1H, furan CH), 6.21 (d, 1H, pyran CH), 5.15 (d, 1H, pyran CH), 3.75 (s, 1H, OH, exchangeable with D2O), 2.75 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3), δ=185.2 (C=O), 163.1 (C-OH), 162.31 (C-O, furan), 152.32 (C-O, pyran), 151.84 (C-N), 134.2, 131.2, 130.5, 129.7, 128.1, 127.9, 127.2, 127.00, 126.5, 126.2, 124.1, 122.5, 119.8, 117.8, 115.8, 112.7, 103.9 (Ar-C), 102.5 (CH, furan), 92.1 (CH-pyran), 42.2 (CH-pyran) 27.2 (CH3). MS (EI, 70 eV): m/z (%)=554 (50%) [M]+. Anal. Calcd. For C32H21F3N2O4 (554.52): C, 69.31; H, 3.82; N, 5.05; Found: C, 69.28; H, 3.79; N, 5.09.
4-(Naphthalen-2-yl)-2-oxo-6-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-3-yl)-1,2-dihydropyridine-3-carbonitrile (15)
A mixture of compound 1 (0.02 mol), compound 2 (0.02 mol), ethyl cyanoacetate (0.02 mol) and ammonium acetate (0.16 mol) in absolute ethanol (50 ml) was refluxed for 10 h. The formed precipitate was filtered, washed with ethanol, dried and recrystallized from acetic acid to give the title compound 15.
Product 15 was separated as yellowish brown crystals, yield 77%. M.p. 220-222°C. IR (KBr): Ṽ =3333 (NH), 2200 (CN), 1675 (C=O), 1643 (C=C), 1564 (N=N), 1230 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.15 (s, 1H, NH, exchangeable with D2O), 8.01 (s, 1H, furan CH), 7.99- 6.53 (m, 15H, Harom); 13C-NMR (125 MHz, CDCl3) δ=160.5 (C=O), 158.6 (C-O), 153.6 (C-N), 133.2, 132.2, 132.5, 128.7, 127.5, 126.4, 124.1, 121.5, 118.8, 117.8, 115.8, 112.2, 103.9 (Ar-C), 113.7 (CN). MS (EI, 70 eV): m/z (%)=534 (35%) [M]+. Anal. Calcd. For C31H17F3N4O2 (534.49): C, 69.66; H, 3.21; N, 10.48; Found: C, 69.69; H, 3.18; N, 10.50.
General procedures for synthesis of 16-18
A mixture of compound 15 (0.004 mol), different alkyl halide namely; methyl iodide, chloroacetone and/or ethyl bromoacetate (0.004 mol) and potassium carbonate (0.56 g, 0.004 mol) in DMF (10 ml) was refluxed for 8-10 h. Then, the reaction mixture was cooled and poured onto ice/cold water. The formed precipitate was filtered, dried and recrystallized from methanol to give the title compounds 16-18, respectively.
2-Methoxy-4-(naphthalen-2-yl)-6-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-3-yl)nicotinonitrile (16): Product 16 was separated as yellowish brown crystals, yield 73%. M.p. 198-199°C. IR (KBr): Ṽ =2200 (CN), 1633 (C=C), 1575 (C=N), 1560 (N=N), 1230 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.03 (s, 1H, furan CH), 8.01-7.53 (m, 15H, Harom), 4.05 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3), δ=165.3 (cyclic C-O), 158.6 (cyclic C-N), 153.6 (C-O, furan), 133.2, 132.2, 132.5, 128.7, 127.5, 126.4, 124.1, 121.5, 118.8, 117.8, 112.2 (Ar-C), 115.7 (CN), 53.2 (CH3). MS (EI, 70 eV): m/z (%)=548 (75%) [M]+. Anal. Calcd. For C32H19F3N4O2 (548.51): C, 70.07; H, 3.49; N, 10.21; Found: C, 70.10; H, 3.45; N, 10.18.
4-(naphthalen-2-yl)-2-(2-oxopropoxy)-6-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-3-yl)nicotinonitrile (17): Product 17 was separated as yellowish brown crystals, yield 62%. M.p. 172-173°C. IR (KBr): Ṽ =2200 (CN), 1735 (C=O), 1633 (C=C), 1575 (C=N), 1560 (N=N), 1230 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.60 (s, 1H, furan CH), 8.33-7.33 (m, 15H, Harom), 5.01 (s, 2H, CH2), 2.05 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3), δ=195.3 (C=O), 165.4 (cyclic C-O), 158.3 (cyclic C-N), 153.2 (C-O, furan), 133.2, 132.2, 132.5, 128.7, 127.5, 126.4, 124.1, 121.5, 118.8, 117.8, 112.2 (Ar-C), 115.7 (CN), 71.5 (CH2), 22.5 (CH3). MS (EI, 70 eV): m/z (%)=590 (65%) [M]+. Anal. Calcd. For C34H21F3N4O3 (590.55): C, 69.15; H, 3.58; N, 9.49; Found: C, 69.18; H, 3.60; N, 9.51.
4-(naphthalen-2-yl)-2-(2-oxobutoxy)-6-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-3-yl)nicotinonitrile (18): Product 18 was separated as yellow crystals, yield 66%. M.p. 132-133°C. IR (KBr): Ṽ =2210 (CN), 1739 (C=O), 1630 (C=C), 1570 (C=N), 1560 (N=N), 1240 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.82 (s, 1H, furan CH), 8.53-6.93 (m, 15H, Harom), 5.04 (s, 2H, CH2), 4.05 (q, 2H, CH2), 1.25 (t, 3H, CH3). 13C NMR (125 MHz, CDCl3), δ=165.3 (C=O), 155.4 (cyclic C-O), 153.3 (cyclic C-N), 150.2 (C-O, furan), 148.1, 133.2, 132.2, 132.5, 128.7, 127.5, 126.4, 124.1, 121.5, 118.8, 117.8, 112.2 (Ar-C), 115.7 (CN), 66.5 (CH2), 61.5 (CH2), 14.5 (CH3). MS (EI, 70 eV): m/z (%)=604 (65%) [M]+. Anal. Calcd. For C35H23F3N4O3 (604.58): C, 69.53; H, 3.83; N, 9.27; Found: C, 69.50; H, 3.80; N, 9.30.
2-((3-cyano-4-(naphthalen-2-yl)-6-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-3-yl)pyridin-2-yl)oxy)acetohydrazide (19)
A mixture of compound 18 (0.004 mol) and hydrazine hydrate (0.008 mol) in absolute ethanol (20 ml) was refluxed for 5 h. The formed precipitate was filtered, dried and recrystallized from ethanol to give the title compound 19.
Product 19 was separated as yellow crystals, yield 60%. M.p. 117-118°C. IR (KBr): Ṽ =3536, 3328 (NH2, NH), 2150 (CN), 1675 (C=O), 1613 (C=C), 1565 (C=N), 1550 (N=N), 1220 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.82 (s, 1H, furan CH), 8.53-6.93 (m, 15H, Harom), 5.04 (s, 2H, CH2), 2.05 (s, 1H, NH, exchangeable with D2O), 1.61 (s, 2H, NH2); 13C-NMR (125 MHz, CDCl3) δ=164.3 (C=O), 154.4 (cyclic C-O), 152.3 (cyclic C-N), 151.2 (C-O, furan), 145.1, 133.2, 132.2, 132.5, 128.7, 127.5, 126.4, 124.1, 121.5, 118.8, 117.8, 112.2 (Ar-C), 114.7 (CN), 65.5 (CH2). MS (EI, 70 eV): m/z (%)=606 (55%) [M]+. Anal. Calcd. For C33H21F3N5O3 (606.55): C, 65.35; H, 3.49; N, 13.86; Found: C, 65.33; H, 3.53; N, 13.88.
General procedures for synthesis of 20a, b
A mixture of compound 19 (0.001 mol) and m and/or p-anisaldehyde (0.001 mol) in absolute ethanol (10 ml) was refluxed for 8 h. After cooling, the formed precipitate was filtered, dried and recrystallized from ethanol to give the title compound 20a, b.
2-((3-cyano-4-(naphthalen-2-yl)-6-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-3-yl)pyridin-2-yl)oxy)-N'-(4- methoxybenzylidene)acetohydrazide (20a): Product 20a was separated as yellow crystals, yield 69%. M.p. 154-155°C. IR (KBr): Ṽ =3348 (NH), 2155 (CN), 1685 (C=O), 1612 (C=C), 1560 (C=N), 1550 (N=N), 1230 (C-F) cm-1. 1H NMR (500 MHz, CDCl3), δ=8.72 (s, 1H, furan CH), 8.63-7.03 (m, 16H, Harom, CH=N), 4.84 (s, 2H, CH2), 3.71 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3), δ=166.3 (C=O), 156.4 (cyclic C-O), 153.3 (cyclic C-N), 151.2 (C-O, furan), 145.1, 133.2, 132.2, 132.5, 128.7, 127.5, 126.4, 124.1, 121.5, 118.8, 117.8, 112.2 (Ar-C), 114.7 (CN), 63.5 (CH2), 56.4 (CH3). MS (EI, 70 eV): m/z (%)=724 (25%) [M]+. Anal. Calcd. For C41H27F3N6O4 (724.69): C, 67.95; H, 3.76; N, 11.60; Found: C, 67.98; H, 3.74; N, 11.58.
2-((3-cyano-4-(naphthalen-2-yl)-6-(5-(3-(trifluoromethyl)phenyl)diazenyl)benzofuran-3-yl)pyridin-2-yl)oxy)-N'-(3- methoxybenzylidene)acetohydrazide (20b): Product 20b was separated as yellow crystals, yield 65%. M.p. 140-141°C. IR (KBr): Ṽ =3348 (NH), 2155 (CN), 1685 (C=O), 1612 (C=C), 1560 (C=N), 1550 (N=N), 1230 (C-F) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.72 (s, 1H, furan CH), 8.63-7.03 (m, 16H, Harom, CH=N), 4.84 (s, 2H, CH2), 3.71 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3), δ=166.3 (C=O), 156.4 (cyclic C-O), 153.3 (cyclic C-N), 151.2 (C-O, furan), 145.1, 133.2, 132.2, 132.5, 128.7, 127.5, 126.4, 124.1, 121.5, 118.8, 117.8, 112.2 (Ar-C), 114.7 (CN), 63.5 (CH2), 56.1 (CH3). MS (EI, 70 eV): m/z (%)=724 (25%) [M]+. Anal. Calcd. For C41H27F3N6O4 (724.69): C, 67.95; H, 3.76; N, 11.60; Found: C, 67.98; H, 3.74; N, 11.58.
Synthesis of 2-((3-cyano-4-(naphthalen-2-yl)-6-(5-((3-(trifluoromethyl)phenyl)diazenyl) benzofuran-3-yl)pyridin-2-yl)oxy)acetyl azide (21)
A stirred carboxyl acid hydrazide 19 (10 mmol) in HCl (20 ml) at 0-5°C, sodium nitrite was added portion-wise till effervesces seated. The reaction mixture was stirred for 1 h. The resulting solid was collected, filtered, washed with water. This solid dried over suction and recrystallized from ethanol to give compound 21.
Product 21 was separated as yellow crystals, yield 72%. M.p. 190-192°C. IR (KBr): Ṽ =2218 (N3), 2180 (CN), 1695 (C=O), 1653 (C=C), 1550 (N=N), 1564 (C=N) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.64 (s, 1H, CH furan), 8.22-7.38 (m, 15H, Harom), 5.26 (s, 2H, CH2); 13C-NMR (125 MHz, CDCl3), δ=163.3 (C=O), 153.4 (cyclic C-O), 150.3 (cyclic C-N), 149.2 (C-O, furan), 144.1, 133.5, 132.3, 132.1, 128.5, 127.1, 126.7, 124.2, 123.5, 119.8, 118.8, 112.2 (Ar-C), 113.7 (CN), 63.5 (CH2). MS (EI, 70 eV): m/z (%)=724 (25%) [M]+. Anal. Calcd. For C33H18F3N7O3 (617.54): C, 64.18; H, 2.94; N, 15.88; Found: C, 64.21; H, 2.90; N, 15.90.
Synthesis of 2-((2,4-dioxothiazolidin-3-yl)methoxy)-4-(naphthalen-2-yl)-6-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-3- yl)nicotinonitrile (22)
A mixture of the azide 21 (1 mmol) and thioglycolic acid (0.12 g, 2 mmol) in 15 ml of dry benzene. The mixture was reflux for 1 h through which the reaction solution was checked with TLC till the end of 21 then concentrated under vacuum, cooled, filtered and crystallized to give compound 22.
Product 22 was separated as yellowish brown crystals, yield 66%. M.p. 163-164°C. IR (KBr): Ṽ =2130 (CN), 1685, 1675 (C=O), 1640 (C=C), 1550 (N=N), 1564 (C=N) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.64 (s, 1H, CH furan), 8.22-7.38 (m, 15H, Harom), 5.26 (s, 2H, CH2), 4.02 (s, 2H, CH2, thiazoldine); 13C-NMR (125 MHz, CDCl3), δ=178.1, 177.4 (2 C=O), 163.3, 158.0 (2 C-O), 151.83 (d, J=1.7 Hz), 148.1, 134.5, 134.0, 132.9, 131.3, 129.7, 124.0, 128.3, 127.9, 126.6, 122.9, 122.7, 121.3, 119.8, 116.6, 114.9, 112.1(Ar-C), 111.5 (CN), 73.9 (CH2), 30.4 (CH2). MS (EI, 70 eV): m/z (%)=663 (50%) [M]+. Anal. Calcd. For C35H20F3N5O4S (663.62): C, 63.35; H, 3.04; N, 10.55; Found: C, 63.31; H, 3.08; N, 10.53.
Synthesis of 2-((2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl)methoxy)-4-(naphthalen-2-yl)-6-(5-((3- (trifluoromethyl)phenyl)diazenyl)benzofuran-3-yl)nicotinonitrile (23)
A mixture of the azide 21 (5 mmol) and anthranilic acid (0.68 g, 5 mmol) in of dry dioxane. The mixture was reflux for 5 h. The resulting solid so formed was collected and crystallized from ethanol to give compound 23.
Product 23 was separated as yellow crystals, yield 79%. M.p. 123-125°C. IR (KBr): Ṽ =3333 (NH), 2118 (CN), 1680, 1670 (C=O), 1635 (C=C), 1560 (C=N) 1550 (N=N) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.56 (s, 1H, CH furan), 8.22-7.23 (m, 19H, Harom), 5.81 (s, 1H, NH, exchangeable with D2O), 5.56 (s, 2H, CH2). MS (EI, 70 eV): m/z (%)=708 (25%) [M]+. Anal. Calcd. For C40H23F3N6O4 (708.64): C, 67.80; H, 3.27; N, 11.86; Found: C, 67.78; H, 3.30; N, 11.85.
General procedures for synthesis of 24a, b
A mixture of the azide 21 (5 mmol) and the appropriate anilines namely, 4-chloroaniline, or 4-aminopyridine (5 mmol), in dry dioxane (20 ml) was refluxed for 5 h. The reaction mixture was evaporated under vacuum the collected solid was crystallized from the proper solvent to give 24a, b.
1-(4-Chlorophenyl)-3-(((3-cyano-4-(naphthalen-2-yl)-6-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-3-yl)pyridin-2- yl)oxy)methyl)urea (24a): Product 24a was separated as yellowish brown crystals, yield 68%. M.p. 179-180°C. IR (KBr): Ṽ =3343, 3323 (NH), 2130 (CN), 1665 (C=O), 1630 (C=C), 1565 (C=N) 1555 (N=N), 512 (C-Cl) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.55 (s, 1H, CH furan), 8.24-7.29 (m, 19H, Harom), 6.24 (s, 1H, NH, exchangeable with D2O), 5.58 (s, 1H, NH, exchangeable with D2O), 5.14 (s, 2H, CH2). MS (EI, 70 eV): m/z (%)=717 (25%) [M]+. Anal. Calcd. For C39H24ClF3N6O3 (717.09): C, 65.32; H, 3.37; N, 11.72; Found: C, 65.35; H, 3.40; N, 11.70.
1-(((3-Cyano-4-(naphthalen-2-yl)-6-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-3-yl)pyridin-2-yl)oxy)methyl)-3-(pyridin-4- yl)urea (24b): Product 24b was separated as yellow crystals, yield 73%. M.p. 194-195°C. IR (KBr): Ṽ =3343, 3322 (NH), 2132 (CN), 1662 (C=O), 1636 (C=C), 1565 (C=N) 1550 (N=N) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.56 (s, 1H, CH furan), 8.55-7.25 (m, 19H, Harom), 6.20 (s, 1H, NH, exchangeable with D2O), 5.40 (s, 1H, NH, exchangeable with D2O), 5.09 (s, 2H, CH2). MS (EI, 70 eV): m/z (%)=683 (25%) [M]+. Anal. Calcd. For C38H24F3N7O3 (683.64): C, 66.76; H, 3.54; N, 14.34; Found: C, 66.77; H, 3.51; N, 14.30.
General procedures for synthesis of 25a, b
A mixture of the azide 21 (5 mmol) and the appropriate phenol namely, phenol, or 4-aminophenol, (5 mmol), in dry benzene (20 ml) was refluxed for 6 h. The reaction mixture was evaporated under vacuum and Petroleum ether was added, the collected solid was crystallized from the proper solvent to give 25a, b.
Phenyl-(((3-cyano-4-(naphthalen-2-yl)-6-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-3-yl)pyridin-2-yl)oxy)methyl)carbamate (25a): Product 25a was separated as reddish brown crystals, yield 75%. M.p. 210-212°C. IR (KBr): Ṽ =3345 (NH), 2135 (CN), 1762 (C=O), 1560 (C=N) 1555 (N=N) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.23 (s, 1H, CH furan), 8.20-7.20 (m, 20H, Harom), 5.09 (s, 2H, CH2), 4.25 (s, 1H, NH, exchangeable with D2O). MS (EI, 70 eV): m/z (%)=683 (25%) [M]+. Anal. Calcd. For C39H24F3N5O4 (683.63): C, 68.52; H, 3.54; N, 10.24; Found: C, 68.50; H, 3.55; N, 10.25.
4-Aminophenyl-(((3-cyano-4-(naphthalen-2-yl)-6-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-3-yl)pyridin-2- yl)oxy)methyl)carbamate (25b): Product 25b was separated as yellowish brown crystals, yield 79%. M.p. 235-236°C. IR (KBr): Ṽ =3542, 3445 (NH, NH2), 2130 (CN), 1760 (C=O), 1560 (C=N) 1555 (N=N) cm-1; 1H-NMR (500 MHz, CDCl3), δ=8.40 (s, 1H, CH furan), 8.36-7.19 (m, 19H, Harom), 5.10 (s, 2H, CH2), 4.25 (s, 1H, NH, exchangeable with D2O), 3.25 (s, 2H, NH2, exchangeable with D2O). MS (EI, 70 eV): m/z (%)=698 (15%) [M]+. Anal. Calcd. For C39H25F3N6O4 (698.65): C, 67.05; H, 3.61; N, 12.03; Found: C, 67.03; H, 3.59; N, 12.00.
Antiviral activity
Material and methods
Cell lines and viruses: HAV stock was kindly provided by Prof. Dr. Celia Barardi, Federal University of Santa Catarina, Barzil. Vero cells, purchased from VACSERA, were grown in presence of Minimum Essential Medium (MEM). The media were contained 10% heat inactivated fetal bovine serum (FBS), 100 μg/ml streptomycin, 100 units/ml penicillin and 1% 4-2-Hydroxyethyl-1-Piperazine Ethane Sulfonic Acid (HEPES) followed by incubation in a humidified 5% CO2 atmosphere. For the cytotoxicity and antiviral assays, the medium contained 2% of fetal bovine serum. Viral stock was prepared and tittered in Vero cells, which assessed from cytopathogenicity of cells caused by viral infection and calculated as 50% Tissue Culture Infectious Doses (TCID50) [30].
Cytotoxicity assay: The cytotoxicity evaluation of the tested compounds was investigated by 3-(4,5-Dimethylthiazol-2-yl)-2,5- Diphenyltetrazolium Bromide (MTT) assay, Briefly, 5 × 104 cells/well were seeded in 96-well plates and incubated in CO2 atmosphere. After 24 h, the growth medium was discarded and the Vero cell monolayers were incubated with various concentrations (ranging from 7.8-1000 μg/ml) of each compound at 37°C under humidified 5% CO2 atmosphere. The compound was removed and 100 μl of MTT (5 mg/ml) solution was added to all wells for 1 h at 37 37°C. The MTT was carefully discarded from wells and replaced with 50 μl DMSO then incubated for 30 min at 37°C. The mean optical density was read using a multi well ELISA reader at 540 nm. The 50% Cytotoxic Concentration (CC50) was estimated as (A-B/ A) × 100, where, A and B are the mean of three OD540 of untreated and treated cells with compounds, respectively.
Study of antiviral activity of compounds on HAV by MTT method: Vero cell lines were seeded in 96- well plate at concentrations of 5 × 104 cells/well and incubated for 24 h. then the culture medium was removed from the wells and the antiviral assays were carried out in three different ways:
Virucidal: 106 log10 TCID50/0.1 ml of HAV was mixed with same volume of various non-toxic doses of each compound and incubated for 1 h at 37°C. One hundred microliters of the above mixture was inoculated onto Vero cells in 96-well plates for 1 h. the mixed solution was discarded from wells and the cell culture were rinsed by test medium and incubated with 200 μl of test medium.
Pre-treatment
Vero cell monolayers grown in 24-well plates were treated with compounds for 24 h at 37°C in 5% CO2 atmosphere. The compound concentrations were removed from the wells followed by infection with 100 μl of 106 log10 TCID50/0.1 of virus. The infected cell monolayers were incubated for 1 h at 37°C, then viral inoculum was discarded. The cell culture was rinsed twice using test medium and incubated with 200 μl of growth medium.
Post-treatment
The experiment was performed as stated above with the following difference: Confluent Vero cell monolayers were infected with 106 log10 TCID50/0.1 of HAV for 1 h. After discarding the viral inoculum, the cells were rinsed using test medium to remove unbound virus then incubated with test medium containing various non-toxic doses of each compound. Controls consisting of virus control (untreated-infected cells) and cell control (untreated non-infected cells) were included in the three protocols. All plates were incubated for 3 days at 37°C in 5% CO2 atmosphere; The 50% inhibitory concentration (IC50) was determined by the MTT method as stated above as [(A-B)/(C-B) × 100] by using multi-well ELISA spectrophotometer at 450 nm, where A, B and C imply to the mean three absorbance of the compound with virus, virus control and cell control, respectively. The Therapeutic Index (TI) was calculated as the ratio of CC50/IC50.
The antiviral activity of compounds on HAV by TCID50/0.1 ml determination
Ten-fold dilutions of HAV were prepared in test medium. 100 μl of each compound was mixed viral dilutions 10-4–10-9, separately. These mixtures were added onto sub confluent cell monolayers in 96-well plates in three different methods as described above with the MTT assay. Microscopic examination for the virus pathogenicity was performed after 3 days post infection. The virus titer as 50% TCID50 was estimated using Spearman Karber method (Finney, 1978). The reduction of virus titer was calculated as the difference between the values of virus with compound and without.
Anticancer activity
Cytotoxic assay (MTT proliferation assay)
The cytotoxic activity of the prepared derivatives was evaluated using MTT proliferation assay. Cells were seeded at 4 × 104 per well in 96-well culture plates before treatment with different concentrations of the tested compound. After treatment for 72 h, the cytotoxicity of the tested compound was determined using the MTT cell proliferation assay (Thiazolyl blue tetrazolium bromide, Sigma-M2128). Light absorbance values (OD=OD570-OD620) were recorded at 570 and 620 nm using an ELISA reader (Anthoslabtec Instrument, Salzburg, Austria) for calculating the concentration that caused 50% inhibition (IC50), i.e., the cell concentration at which the light absorbance value of the experimental group is half that of the control group. These results were expressed as a percentage of the control ± SD established from n=4 wells per experiment from three independent experiments.
Results and Discussions
Chemistry
The synthetic strategy that reported herein depends on the key synthon; 3-(Naphthalen-2-yl)-1-(5-((3- (trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)prop-2-en-1-one (3), that synthesized according the known method (Claisen-Schmidt condensation) of addition of equimolar amount of acetyl benzofuran derivative 1 and naphthyl-4-carboxaldehyde 2 in alcoholic sodium hydroxide (10%, 200 ml) to stir overnight at room temperature. Compound 3 showed a strong band at 1655 cm-1 at its IR spectrum because of α, β-unsaturated ketone. All synthesized compounds according to the synthetic strategy are depicted in Schemes 1-4.
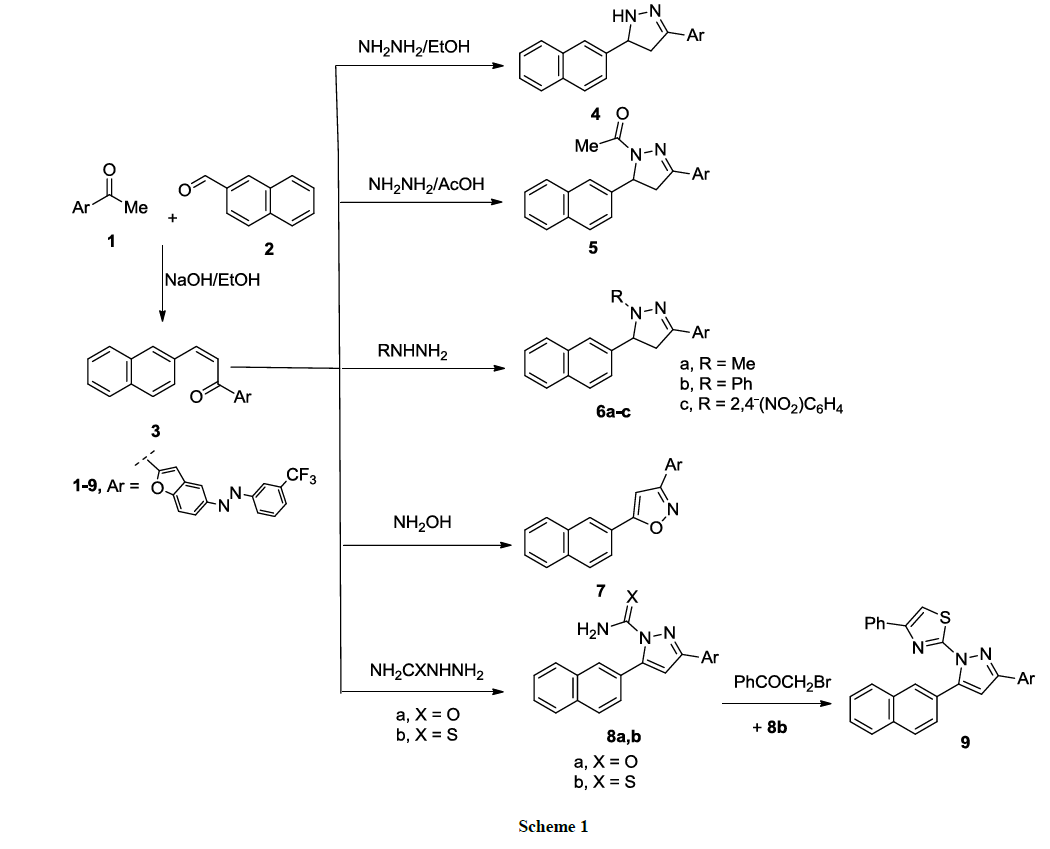
Scheme 1
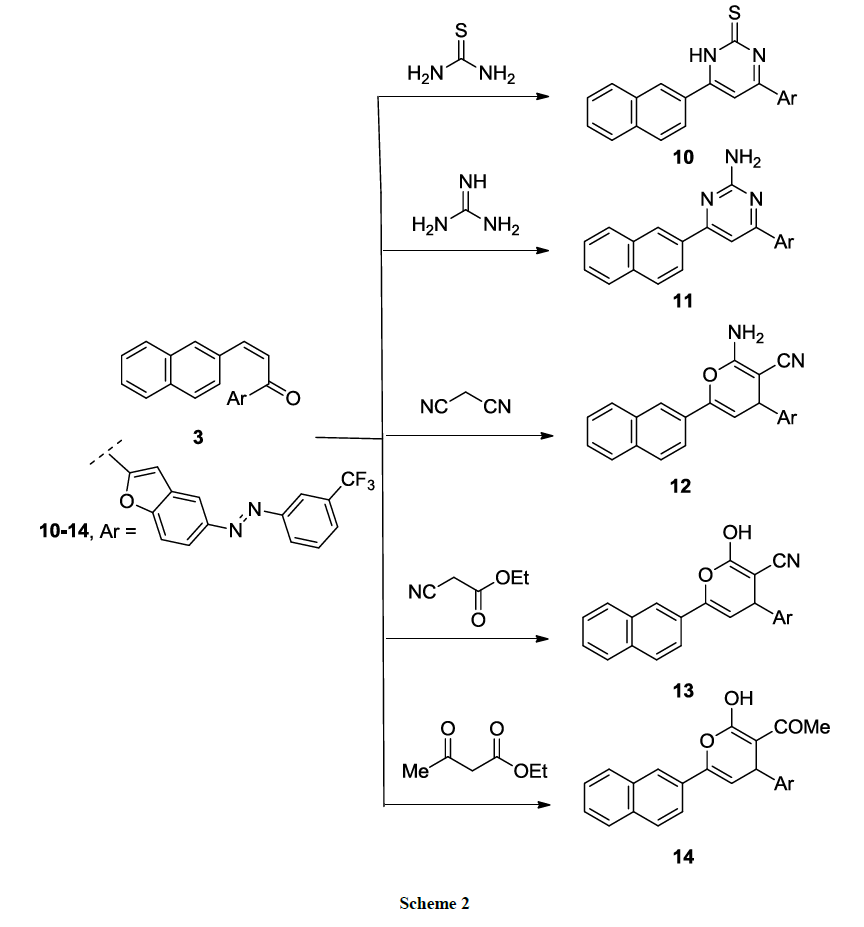
Scheme 2
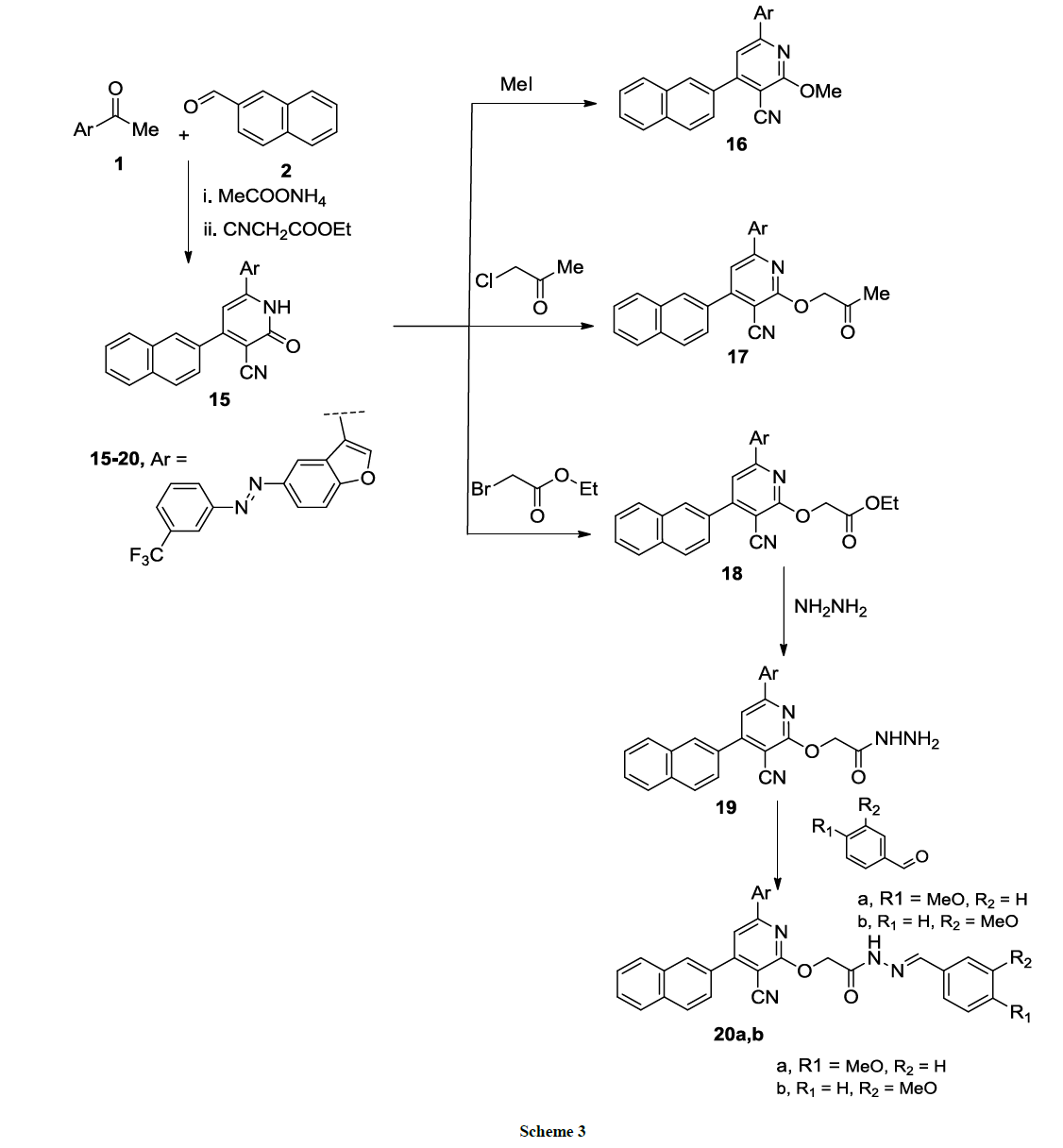
Scheme 3
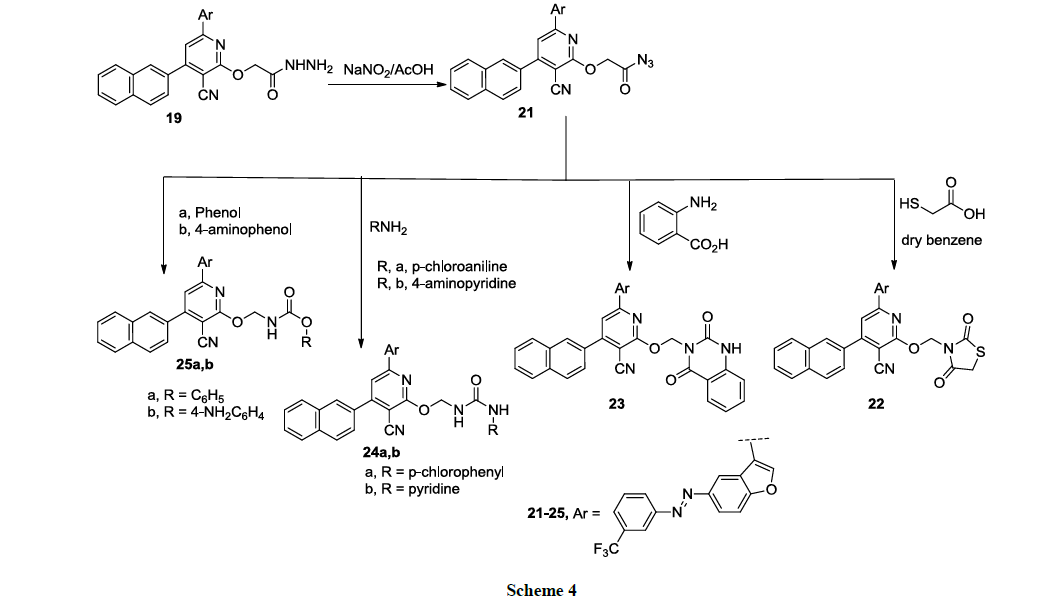
Scheme 4
Cyclocondensation of compound 3 with hydrazine hydrate derivatives namely, hydrazine hydrate, methyl hydrazine, phenyl hydrazine and 2,4- dinitrophenyl hydrazine, it gave pyrazoline derivatives 4, 5, 6a-c (Scheme 1). The most characteristic feature of these pyrazoline derivatives is the absence of α, β-unsaturated ketone absorption band in their IR spectra (cf Experimental).
1H-NMR spectra of compounds 4, 5, 6a-c showed the signals of Ha, Hb, Hx of pyrazoline ring as doublet of doublet in the regions 2.66-3.15 ppm, 2.90-3.35 ppm and 3.72-5.12 ppm, respectively.
On the other hand, in alcoholic sodium hydroxide, cyclization of the chalcone 3 with hydroxylamine hydrochloride at reflux temperature was done; afforded (5-(naphthalen-2-yl)-3-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)isoxazole (7). Its mass spectrum showed a peak at 483 referring to the molecular ion peak (Scheme 1).
Finally, the chalcone 3 reacted with semicarbazide or thiosemicarbazide in absolute ethanol in presence of sodium acetate at reflux temperature; afforded the pyrazole derivative 8a, b in a good yield (Scheme 1). Pyrazole-1-carbothioamide derivative 8b reacted with phenacyl bromide in absolute ethanol to give the corresponding 2-(5-(naphthalen-2-yl)-3-(5-((3-(trifluoromethyl)phenyl)diazenyl)benzofuran-2-yl)-1H-pyrazol-1-yl)- 4-phenylthiazole (9) (Scheme 1). The structures of all synthesized compounds were confirmed based on elemental and spectroscopic analyses (cf. Experimental).
Moreover, thiopyrimidine 10 and amino pyrimidine 11 derivatives were synthesized via cyclocondensation of chalcone 3 with thiourea and guanidine sulfate in sodium ethoxide solution respectively, at reflux temperature (Scheme 2). While pyrane derivatives 12-14 were yielded in heating of chalcone 3 with active methylene compounds namely; malononitrile, ethyl cyanoacetate or ethyl acetoacetate in sodium ethoxide solution (Scheme 2). IR spectrum of pyrane derivative 12 showed presence of absorption band of CN at 2150 cm-1 and NH2 absorption band at 3571 cm-1 while IR spectrum of derivative 14 showed carbonyl C=O absorption band at 1710 cm-1 and OH absorption band at 3215 cm-1. The structures of compounds 10-14 were confirmed on the bases of elemental and spectroscopic analyses (cf. Experimental).
Pyridine-3-carbonitrile derivative 15 could be successfully synthesized at one-pot reaction of acetyl benzofuran 1, naphthyl-4-carboxaldehyde 2, ethyl cyanoacetate and ammonium acetate in absolute ethanol (Scheme 3). IR spectrum of compound 15 showed absorption bands at 3333 (NH), 2200 (CN), 1675 (C=O) cm-1. 1H-NMR spectrum showed singlet peak at δ=8.15 of (NH) which is exchangeable with D2O. The derivative 15 reacted with different alkyl halide namely; methyl iodide, chloroacetone or ethyl bromoacetate in dry DMF containing anhydrous K2CO3 yielded the corresponding 2- pyridine derivatives 16-18, respectively (Scheme 3).
1H-NMR spectrum of derivative 61 had a singlet signal at δ=4.05 ppm of CH3 group and compound 17 showed two singlet signals at δ=5.01 ppm (CH2) and δ=2.05 ppm (CH3); while 1H-NMR spectrum of compound 18 showed a singlet signal at δ=5.04 (CH2), δ=4.05 (q, 2H, CH2), δ=1.25 (t, 3H, CH3).
Compound 18 reacted with hydrazine hydrate in absolute ethanol to give the hydrazide derivative 19 in good yield (Scheme 3) which condensed with m, or p-anisaldehyde to give the corresponding Schiff base derivatives 20a, b (Scheme 3). The structures of compounds 16-20a, b were assigned according to their analytical and spectroscopic analysis (cf. Experimental).
The reaction of 19 with sodium nitrite in acetic acid at 2-5°C in ice bath, afforded the corresponding carbonyl azide 21, which was further reacted with thioglycolic acid in dry benzene to provide the corresponding thiazolidinedione derivative 22 (Scheme 4).
The reaction of the carbonyl azide 19 with anthranilic acid in dry dioxane afforded the diquinazolinone derivative 23. The reaction of the carbonyl azide 19 with different aniline derivatives namely, p-chloroaniline, 4-aminopyridine in dry dioxane, provided the corresponding substituted urea derivatives 24a, b, respectively (Scheme 4). Finally, The carbamate derivatives 25a, b were obtained upon the reaction of the carbonyl azide 19 with phenols namely, phenol, or 4-aminophenol, in dry benzene (Scheme 4). Structures of new compounds 19-25a, b were illustrated by chemical microanalyses and spectroscopic analyses, IR, 1H-NMR and mass spectra (cf. Experimental).
Biological evaluation: Antiviral activity
Hepatitis A Virus (HAV) infect the liver and primarily transmitted through the faecal-oral route, either by consumption of contaminated water or food or by person-to-person contact. Also, HAV can be transmitted sexually among men who have sex with men. In addition, infected syringes or blood components can help in HAV transmission [31,32]. HAV belongs to the genus Hepatovirus within the Picornaviridae family. HAV is a single strand of RNA covered by icosahedral shaped protein shell [33-35]. HAV is the most common pathogen of clinically apparent viral hepatitis. Approximately 1.4 million new infections by HAV are estimated to occur each year globally, with 11%-22% of cases requiring hospitalization [36-38].
Before performing the antiviral test against Hepatitis A virus, toxicological effects of 15 compounds were investigated by MTT proliferation method. The results of cytotoxicity on cell lines were different depending on compound and CC50 values ranged from 248 μg/ml to 1359 μg/ml. For antiviral assay, three non-toxic doses from each compound have been selected to antiviral experiments by MTT and TCID50 assays. The antiviral assays were carried out in three different protocols to assess whether the compound inhibits viral capsid, receptor, or life cycle.
Our results (Table 1) suggested that all compounds have antiviral activity HAV with therapeutic index ranged from 0 to 4.75 log10 TCID50. Compound 14 showed the maximum inhibitory effect against virus infection, reducing the virus titers by 4.75 log10 TCID50, when it was added to cells before or after entry into host cells. The same efficacy was observed from compound 25a, reducing the virus titers by 4.75 log10 TCID50, but when they incubated with virus before infection. Mechanistically, this compound may be inhibited viral replication by interacting with virus capsid and prevented its binding to viral receptor on cell surface. Also, compound 12 showed strong antiviral activity against HAV, reducing the virus titers by 3.75 log10 TCID50, when it incubated with virus before infection. In addition, moderate inhibitory effect against HAV infections were shown from compounds 3, 16, 20b, 21, 22 and 24b; reducing the virus titers by 1-2 log10 TCID50, whereas compounds 5, 6c, 10, 15, 19 and 23 showed the lowest antiviral activity against HAV infection; reducing the virus titers less than 1 log10 TCID50.
| Compounds | CC50 (μg/ml)a | Virucidal | Treatment before infection | Treatment after infection | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 | TIc | R | IC50 | TIc | R | IC50 | TIc | R | ||
| 3 | 452.9 | 433 | 1 | 0.25 | 781.5 | 0.6 | 0 | 23 | 19.7 | 1 |
| 5 | 578 | 231 | 2.5 | 0.25 | 190 | 3 | 0.25 | 52 | 11 | 0.75 |
| 6c | 157.2 | 203 | 0.8 | 0 | 267 | 0.6 | 0 | 47 | 3.3 | 0.25 |
| 10 | 1486 | 248 | 7 | 0.5 | 238 | 6 | 0.5 | 135 | 11 | 0.75 |
| 12 | 248 | 4.5 | 55 | 3.75 | 29 | 8.5 | 0.5 | 42 | 30.2 | 1.5 |
| 14 | 987.5 | 124 | 8 | 0.5 | 15 | 65.8 | 4.5 | 14 | 70.5 | 4.75 |
| 15 | 462 | 15 | 30.8 | 1.5 | 25 | 18.5 | 1 | 48 | 9.6 | 0.5 |
| 16 | 762.8 | 89 | 8.6 | 0.5 | 145 | 5.3 | 0.5 | 20 | 38 | 2.5 |
| 19 | 537.6 | 71 | 7.6 | 0.5 | 258 | 2 | 0.25 | 42 | 12.8 | 0.75 |
| 20b | 688 | 12 | 57.3 | 4 | 45.5 | 15 | 0.75 | 41.6 | 16.5 | 1 |
| 21 | 985 | 54 | 18.2 | 1 | 75 | 13 | 0.75 | 30 | 32.8 | 2 |
| 22 | 681.5 | 898 | 0.7 | 0 | 988.5 | 0.7 | 0 | 23 | 29.6 | 1.5 |
| 23 | 1359 | 899 | 1.5 | 0.25 | 71 | 19 | 1 | 899 | 1.5 | 0.25 |
| 24b | 258.6 | 222 | 1.2 | 0.25 | 24 | 10.8 | 0.5 | 12 | 21.5 | 1.25 |
| 25a | 746.2 | 11 | 68 | 4.75 | 89.7 | 8.3 | 0.5 | 521 | 1.4 | 0.25 |
CC50: the concentration of extract required to kill 50% of viable cells; R: Reduction of virus titer was calculated as the difference between treated and untreated virus; aConcentration of extract that is cytotoxic to 50% of cells; bConcentration of extract that inhibits viral infectivity (Cytopathic Effect) by 50%; cTherapeutic index=CC50/IC50=The mean values of triplicate experiments; RReduction in virus titer calculated as the difference between treated and untreated virus and expressed in log10 TCID50/0.1.
Table 1: The cytotoxic and anti-HAV of compounds with the mode of action on vero cells determined by MTT method and TCID50/0.1
Biological evaluation: Cytotoxic activity
Cancer remains one of the major life threatening ailments all over the world [39]. It is expected that the number of patients diagnosed with cancer will double by 2050 [40]. Finding new cytotoxic agents is crucial element in the continuous war between humanity and cancer. Scientists try to find more potent cytotoxic agents with fewer side effects by preparing new chemical derivatives of certain scaffolds and evaluating their cytotoxic activity. Certain chemical scaffolds are considered privileged structures due to their unique electronic and steric environment as well as their wide array of biological activities.
Seven of the newly synthesized compounds were selected as representative examples to evaluate their in vitro cytotoxic activity. The obtained cytotoxic results indicates that the new substituted naphthalene derivatives are non-promising antitumor agents against U937, MOLT-4 and K562 except compounds 14 and 15 against MOLT-4 and compound 21 against U937, MOLT-4 and K562 (Table 2).
| Compound | U937 (IC50 mg/ml) |
MOLT-4 (IC50 mg/ml) |
IC50 (IC50 mg/ml) |
|---|---|---|---|
| 3 | NA | NA | NA |
| 12 | NA | NA | NA |
| 14 | NA | 16.91 | NA |
| 15 | NA | 10.92 | NA |
| 20b | NA | NA | NA |
| 21 | 38.9 | 16.08 | 31.15 |
| 25a | NA | 35.05 | NA |
IC50: Dose of the compounds, which reduces survival to 50%; NA: Not active at 40 μg/ml for 72 h; U937: Cell line human lymphoblast lung from human; MOLT-4 (Human T lymphoblast; Acute lymphoblastic leukemia) cell line slides; K562: Cell line human leukemic blood from human
Table 2: Effect of some selected newly synthesized compounds on U937, MOLT-4 and K562 carcinoma cell line
Acknowledgement
The authors thank the National Research Centre for the financial support (Project number 11010317). Also, The authors are grateful to Professor Yu-Cheng Chen, The PhD Program of Cancer Biology and Drug discovery, China Medical University, Taichung, Taiwan and Professor Mei- Chin Lu; Graduate Institute of Marine Biology, National Dong Hwa University, Pingtung, 944, Taiwan; National Museum of Marine Biology & Aquarium, Pingtung 944, Taiwan; for antitumor evaluation.
References
- S. Maayan, N. Ohad, K Soliman, Bioorg. Med. Chem., 2005, 13(2), 433-441.
- Z. Nowakowska, Eur. J. Med. Chem., 2007, 42(2), 125-137.
- M.L. Go, X. Wu, X.L. Liu, Curr. Med. Chem., 2005, 12(4), 481-499.
- S.H.R. Kumar, K.R. Kapoor, K.A. Bansal, S. Khurana, S. Singh, J. Thomas, B. Roy R. Phartyal, S. Saluja, S. Kumar, Anticancer Agents Med Chem., 2015, 16(2), 200-211.
- S. Shenvi, K. Kumar, K.S. Hatti, K. Rijesh, L. Diwakar, G. Chandrasekara Reddy, Eur. J. Med. Chem., 2013, 62, 435-442.
- C.W. Mai, M. Yaeghoobi, N. Abd-Rahman, Y.B. Kang, M.R. Pichika, Eur. J. Med. Chem., 2014, 77, 378-387.
- A. Voskiene, V. Mickevicius, G. Mikulskiene, ARKIVOC., 2007, 15, 303-314.
- C. Viegas-Junior, A. Danuello, V. da Silva Bolzani, E.J. Barreiro C.A.M. Fraga, Curr. Med. Chem., 2007, 14, 1829-1852.
- J.J. Walsh, A. Bell, Curr. Pharm. Des., 2009, 15, 2970-2985.
- N. Anand, P. Singh, A. Sharma, S. Tiwari, V. Singh, D.K. Singh, K.K. Srivastava, B.N. Singh, Bioorg. Med. Chem., 2012, 20, 5150-5163.
- C. Karthikeyan, V.R. Solomon, H. Lee, P. Trivedi, Biomed. Preven. Nutr., 2013, 3, 325-330.
- K.J. Ryan, C.G. Ray (Editors), Sherris Medical Microbiology, 4th Edi.,). McGraw Hill, 2004, 541-544.
- F.E. André, Curr. Top. Microbiol. Immunol., 2006, 304, 95-114.
- F. Cui, S.C. Hadler, H. Zheng, F. Wang, W. Zhenhua, H. Yuansheng, X. Gong, Y. Chen, X. Liang, J. Epidermal., 2009, 19(4), 189-195.
- D. Daniels, S. Grytdal, A. Wasley, MMWR Surveill Summ., 2007, 58(3), 1-27.
- M. Barker, Clackers, M. Copley, R. Demaine, D.A. Inglis, G.G.A. Johnston, M.J. Jones, H.T. Haas, M.V. House, R. Loiseau, J. Med. Chem., 2006, 49, 4216-4231.
- E.S. Al-Abdullah, Molecules., 2011, 16, 3410-3419.
- N.A. Hamdy, A.M. Gamal-Eldeen, H.A. Abdel-Aziz,, I.M.I. Fakhr, Eur. J. Med. Chem., 2010, 45, 463-470.
- Z. Ates-Alagoz, S. Yildiz, E. Buyukbingol, Chemotherapy., 2007, 53, 110-113.
- H. Tang, Y.J. Zhou, Y.W. Li, J.G. Lv, C.H. Zheng, J. Chen, J. Zhu, Chin. Chem. Lett., 2008, 19, 264-268.
- M.S. Al-Mutairi, E.S. Al-Abdullah, M.E. Haiba, M.A. Khedr, W.A. Zaghary, Molecules., 2012, 17, 4717-4732.
- I. Koca, A. Ozgur, K.A. Coskun, Y. Tutar, Bioorg. Med. Chem., 2013, 21, 3859-3865.
- K.M. Dawood, T.M.A. Eldebss, H.S.A. El-Zahabi, H. Mahmoud, M.H. Yousef, P. Metz, Eur. J. Med. Chem., 2013, 70, 740-749.
- I. Bildirici, A. Sener, I. Tozlu, Med. Chem. Res., 2007, 16, 418.
- C.B. Sangani, J.A. Makawana, X. Zhang, B. Shashikant, S.B. Teraiya, L. Lin, H.L. Zhu, Eur. J. Med. Chem., 2014, 76, 549-557.
- S. Mert, R. Kasımogulları, T. Iça, F. Çolak, A. Altun, S. Ok, Eur. J. Med. Chem., 2014, 78, 86-96.
- M.A. Ilies, B. Masereel, S. Rolin, A. Scozzafava, G. Campeanu, V. Cimpeanu, C.T. Supuran, Bioorg. Med. Chem., 2004, 12, 2717-2726.
- I.F. Angelillo, C.G. Nobile, F. Talarico, M. Pavia, Infection.,1996, 24(2), 147-50.
- M.S. Abd-Rabou, Synthesis and Anticancer Screening of Novel Series of Tetrahydronaphthalene incorporated into Different Heterocycles, MSc thesis, Faculty of Pharmacy, Zagazig University, Egypt, 2016.
- M.M. Saed Wassel, Synthesis of some Triazole and pyrazole Derivatives Containing Adamantyl Moiety with expected biological Activity, MSc thesis, Faculty of Science, Cairo University, Egypt, 2015.
- D.J. Finney, Assays based on quantal responses. Statistical Method in Biological Assays (3rd Edn). Macmillan Publishing Co., Inc., NY, USA, 1978, 394-398.
- F. Ansaldi, B. Bruzzone, M.C. Rota, A. Bella, M. Ciofi degli Atti, P. Durando, Eur. J. Epidemiol., 2008, 23(1), 45-53.
- B.D. Schoub Hepatitis, In: M.D. Miliotis, J.W. Bier (Eds) International Handbook of Foodborne Pathogens. Marcel Dekker, New York, 2003, 15-25.
- N. Cook, A. Rzezutka, In: Y. Motarjemi, M. Adams (Eds) Emerging Foodborne Pathogens, Wood head Publishing, Cambridge, 2006, 282-308.
- F. Rozenberg, C. Deback, H. Agut, Infectious Disorders-Drug Targets., 2011, 11, 235-250.
- J.A. Cuthbert, Clin. Microbiol. Rev., 2001, 14 38-58.
- A. Martin, S.M. Lemon, Hepatology., 2006, 43, S164-S172.
- R.M. Pinto, L. D'Andrea, F.J. Perez-Rodriguez, M.I. Costafreda, E. Ribes, S. Guix, A. Bosch, Future Microbiol., 2012, 7, 331-346.
- M.B. Gu¨rdere, O.G, Ayse Sahin Yaglioglu, Y. Budak, M. Ceylan, Res. Chem. Intermed., 2017, 43, 1277-1289.
- E. Sonnenschein, J.A. Brody, J. Gerontol. B. Psychol. Sci. Soc. Sci., 2005, 62(2), S110-S112.



