Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 8
Synthesis, Anti-HIV and Anti-Microbial Activity of Substituted 1-(5-(4-Phenyl)-2-Mercapto-1H-Imidazol-1-Yl)-3-Phenylthiourea Derivatives
Rohit Singh* and Swastika Ganguly
Department of Pharmaceutical Sciences, Birla Institute of Technology, Mesra, Ranchi-835215, Jharkhand, India
- *Corresponding Author:
- Rohit Singh
Department of Pharmaceutical Sciences
Birla Institute of Technology
Mesra, Ranchi-835215, Jharkhand, India
Abstract
In the current study, a series of five 1-phenyl-3-(5-phenyl-1H-imidazol-1-yl) thiourea analogs (5a-5e) have been synthesized, characterized by physical (Melting point, Thin Layer Chromatography) and spectral data (IR, 1H-NMR, and Mass spectrometry) and evaluated for their AntiHIV, Antibacterial, Antifungal and Anthelmintic activity with the aim to develop novel imidazole analogs with broad-spectrum chemotherapeutic properties. Compounds 5a and 5b being the most active showed therapeutic index that was 17.5 and 19.9 compared to Zidovudine (AZT) having the Therapeutic Index (TI) 514342.6. On the other hand, it shows an excellent antibacterial, antifungal as well as anthelmintic activity.
Keywords
Imidazole derivatives, Reverse transcription, Anti-HIV, Anti-bacterial activity, Antifungal activity, Anthelmintic activity.
Introduction
Human immunodeficiency virus type 1 (HIV-1) which is a major cause of AIDS and is the cause of death worldwide. From the data of World Health Organization (WHO) AIDS Epidemic Update at the end of 2016, 36.7 million deaths have been listed out, in addition to 70 million people currently living with AIDS [1]. HIV-1 Reverse transcriptase inhibitors are classified into two different groups: nucleoside analog reverse-transcriptase inhibitors (NRTIs), and non-nucleoside analog reverse-transcriptase inhibitors (NNRTIs) [2]. Edward et al [3] synthesized a series of 5-aryloxysubstituted imidazole derivatives 1 (Figure 1). Interestingly, the S-linked imidazole derivatives have generated good interest in the anti-HIV area, exemplified by capravirine, which exhibits better potency profile against reverse transcription activity [4]. They also showed corresponding O-linked imidazole derivatives shown in Figure 1 would retain the desirable mutant potency profile of compounds such as capravirine.
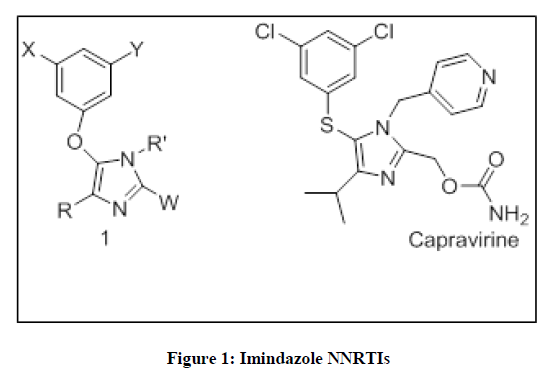
Figure 1: Imindazole NNRTIs
Previous work of our laboratory has been recognized as various substituted imidazole analogs showing broad-spectrum chemotherapeutic activities. In continuation of this work our sincere effort to develop a variety of newer imidazole (Figure 2) analogs as broad-spectrum chemotherapeutic agents, the present study was undertaken to synthesize and evaluate novel imidazoles for antibacterial and antifungal activities [5]. In various oxidation states imidazole has shown a number of interesting biological activities, like antiviral [6,7], antibacterial [8] antifungal [9,10], antiprotozoal [11-13], antihypertensive [14], antihistaminic [15], alpha-adrenergic agonist [16], and blocking [17] and others [18-19].
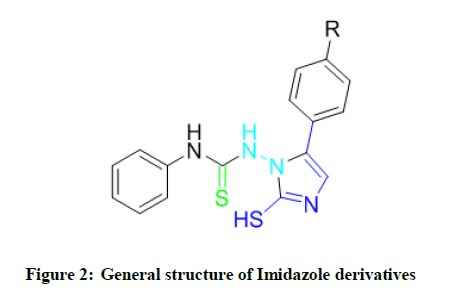
Figure 2: General structure of Imidazole derivatives
Material and Methods
Material
All the reagents and solvents were purchased from local commercial suppliers like Sigma Aldrich, Merck India Ltd., and Rankem chemicals. All reagents were Guaranteed Reagent (GR) or Analytical Reagent (AR) grade and were used without further purification. The purity of the compounds was assessed by the Thin Layer Chromatography technique (TLC) performed on aluminum sheets of silica gel G purchased from Merck by using chloroform: methanol (9: 1) as eluent. For visualization of TLC spots of synthesized compound Iodine chamber and Shimadzu (UV-254) spectrometer were used. Ashless Whatman filter paper was used for vacuum filtration. Melting points (Opti Melt) an automatic apparatus were used for determination of melting point of compound and were uncorrected. IR spectra (KBr disc/pellets) were recorded on SHIMADZU FT-IR 8400 and reported in cm−1. 1H-NMR spectra were recorded at 400 MHz with BRUKER. Chemical shifts are expressed in parts per million (ppm) δ-values relative to Tri methyl silane (TMS) as an internal standard as a reference, using DMSO/CDCl3 as the solvent.
Methods of synthesis
General procedure
Synthesis of ammonium phenyl carbamodithioate: Various Mono or Di substituted anilines (0.071 mol) were added to the mixture of carbon-di-sulfide (0.071 mol) and concentrate Ammonia (0.52 mol) slowly drop wise with vigorous stirring for 60 min. A milky homogenous mixture was formed initially. Keep it an ice bath for 2 h to yield a solid mass. A precipitate was formed as powder, filtered the content and washed with ether solution and dried.
Conversion of ammonium phenyl carbamodithioate to methyl phenyl carbamodithioate: Ammonium phenyl carbamodithioate was dissolved in 10 ml of methanol with cooling in an ice bath. Methyl iodide (0.01 mol) was added to a solution and stirred vigorously at temperature 0ºC-4ºC for 3 h.
Conversion of methyl phenyl carbamodithioate to phenyl thiosemicarbazide: Intermediate as methyl phenyl carbamodithioate was dissolved in 15 ml of methanol at 60ºC. 85% hydrazine hydrate (0.025 mol) was added to the reaction mixture and stirred regularly for 2 h. A reaction mixture was cooled down at room temperature to yield crystalline solid mass.
Synthesis of para substituted phenyl-2-mercapto-1-H-imidazol-1-yl-3-phenyl thiourea: In a reaction mixture of para substituted phenacyl bromide (0.002 mol) in glacial acetic acid (10 ml), Potassium thiocyanate (0.002 mol) was added to the reaction mixture with continuous stirring at the temperature of 60ºC. The reaction mixture was then stirred for further 30 min at same temperature. The intermediate as phenyl thio semicarbazide was added to reaction mixture and stirring was continued at 60ºC for 1 h and the reaction mixture then refluxed for 6 h. When reflux was over, 30 ml of water was added to the reaction mixture which yielded a gummy mass. Further subsequent washing was carried out with ether (25 × 10 ml) and product was precipitated out as solid mass which was further re-crystallized with ethanol-water mixture. The substituent of final product is shown in Table 1.
| S. No. | -R |
|---|---|
| 5a | -Cl |
| 5b | -Br |
| 5c | 0 |
| 5d | -OCH3 |
| 5e | -C6H5 |
Table 1: Substituent at R
1-(5-(4-chlorophenyl)-2-mercapto-1H-imidazol-1-yl)-3-phenylthiourea (5a) (Figures 3 and 4)
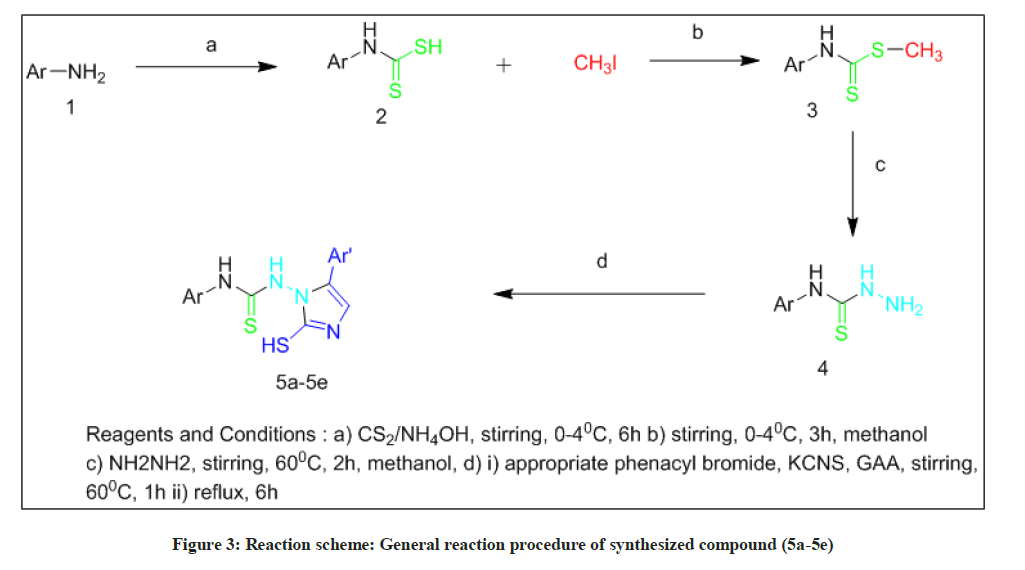
Figure 3: Reaction scheme: General reaction procedure of synthesized compound (5a-5e)
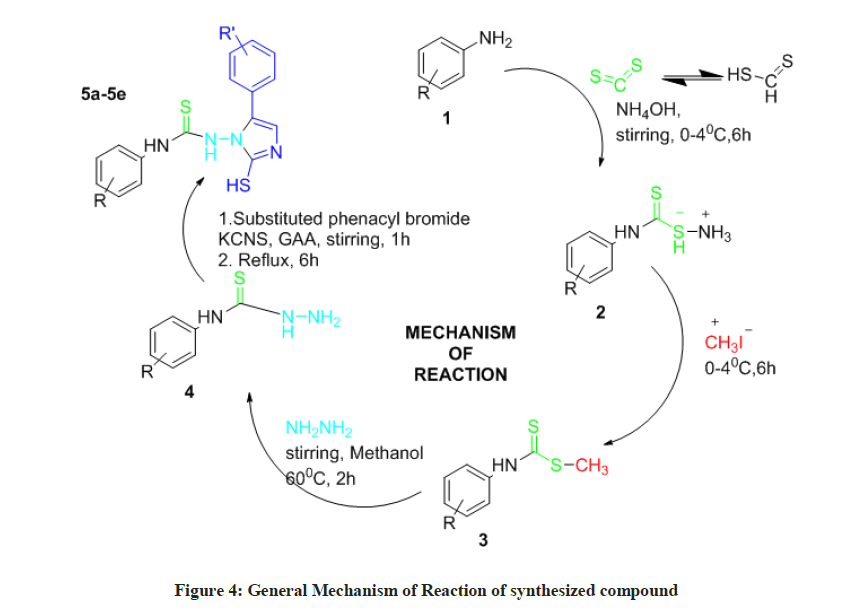
Figure 4: General Mechanism of Reaction of synthesized compound
Yield: 76%, M.P. 80oC-82oC, IR (KBr, cm-1): 3058 (N-H stretching), 2057 (C-H stretch CH3 group) 1599 (C=S stretching), 1314 (C-N stretching) cm-1; NMR (DMSO, 400 MHz): 2.65 (s; 1H; SH), 7.92 (s; 1H; NH-Ar), 7.60-7.07 (m; 9H; Ar-H), 4.69 (s; 1H; NH-imidazole), EI MS: 359.04 [M+1].
1-(5-(4-bromophenyl)-2-mercapto-1H-imidazol-1-yl)-3-phenylthiourea (5b)
Yield: 65%, M.P. 88oC-90oC, IR (KBr, cm-1): 3269 (N-H stretching), 2359 (C-H stretch CH3 group) 1659 (C=S stretching), 1405 (C-N stretching) cm-1; NMR (DMSO, 400 MHz): 2.66 (s; 1H; SH), 7.67 (s; 1H; NH-Ar), 7.66-7.09 (m; 9H; Ar-H), 4.68 (s; 1H; NH-imidazole), 402.99 [M+1].
1-(2-mercapto-5-p-tolyl-1H-imidazol-1-yl)-3-phenylthiourea (5c)
Yield: 79%, M.P. 66oC-68oC, IR (KBr, cm-1): 3303 (N-H stretching), 2924 (C-H stretch CH3 group) 1608 (C=S stretching), 1311 (C-N stretching) cm-1; NMR (DMSO, 400 MHz): 2.44 (s; 3H; CH3) 2.64 (s; 1H; SH), 7.92 (s; 1H; NH-Ar), 7.60-7.07 (m; 9H; Ar-H), 4.73 (s; 1H; NH-imidazole), 338.10 [M+1].
1-(2-mercapto-5-(4-methoxyphenyl)-1H-imidazol-1-yl)-3-phenylthiourea (5d)
Yield: 67%, M.P. 71oC-73oC, IR (KBr, cm-1): 3237 (N-H stretching), 2364 (C-H stretch CH3 group) 1670 (C=S stretching), 1301 (C-N stretching) cm-1; NMR (DMSO, 400 MHz): 2.25 (s; 1H; SH), 7.96 (s; 1H; NH-Ar), 7.57-6.91 (m; 9H; Ar-H), 4.70 (s; 1H; NH-imidazole), 354.09 [M+1].
1-(2-([1,1'-biphenyl]-4-yl)-5-mercaptocyclopenta-2,4-dien-1-yl)-3-phenylthiourea (5e)
Yield: 82%, M.P. 101oC-103oC, IR (KBr, cm-1): 3026 (N-H stretching), 2359 (C-H stretch CH3 group) 1660 (C=S stretching), 1252 (C-N stretching) cm-1; NMR (DMSO, 400 MHz): 2.66 (s; 1H; SH), 7.78 (s; 1H; NH-Ar), 7.65-6.71 (m; 14H; Ar-H), 4.77 (s; 1H; NH-imidazole), 401.11 [M+1].
Anti-Hiv activity
Sample and chemicals
All testing samples were provided by Dr. Swastika Ganguly (Department of Pharmaceutical Science and Technology, BIT, Mesra, Jharkhand, India). All provided samples were dissolved in DMSO. Zidovudine (AZT) was purchased from USP and dissolved in the serum-free RPMI-1640 medium.
Reagents
HEPES (N-2 (2-Hydroxyethyl) piperazine-N'-(2-ethane sulfonic acid), MTT (3,(4,5-dimethylthiazol-2-yl) -2,5-diphenyl tetrazolium bromide), DMF (N, N'- Dimethyl formamide), Penicillin, Streptomycin sulfate, Glutamine were purchased from Sigma; 2-ME (2-Mercaptoethanol) was purchased from Bio-Rad. RPMI-1640 and fetal bovine serum (FBS) were purchased from Gibco.
Cells and viruses
C8166 cell and HIV-1IIIB were kindheartedly donated by Medical Research Council, AIDS Regent Project. The cells were preserved at 37ºC in 5% CO2 in RPMI-1640 medium along with 10% heat-inactivating FBS (Gibco). HIV-1IIIB was prepared from the supernatants of H9/HIV-1IIIB cells. The 50% HIV-1 tissue culture infectious dose (TCID50) in C8166 cells was resolute and calculated by Reed and Muench method. Virus stocks were stored in small aliquots at -70ºC. The titer of virus stock was 1.0 × 108 TCID50 per ml.
Cytotoxic assay
The cellular toxicity of synthesized compounds on C8166 was assessed by MTT colorimetric assay. Briefly, 100 μl of 4 × 105 cells were plated into 96 well plates. 100 μl of various concentrations of compounds were added and incubated at 37ºC in a humidified atmosphere of 5% CO2 for 72 h. Discard 100 μl supernatant, MTT reagent was added and incubated for 4 h, 100 μl 50% DMF-15% SDS was added. After the formazan was dissolved completely, the plates were analyzed by a Bio-Tek ELx 800 ELISA reader at 570 nm/630 nm. 50% cytotoxicity concentration (CC50) was calculated.
Inhibition of syncytia formation
The inhibition effect of samples on acute HIV-1 infection was observed by the syncytia formation assay. In the presence or absence of various concentrations of samples, 4 × 104 C8166 cells were infected with HIV-1 at a multiplicity of infection (MOI) of 0.04, and cultured in 96-well plates at 37ºC in 5% CO2 for 3 days. Efavirenz was used as a positive control. At 3 days post-infection, Cytopathic effect (CPE) was measured by counting the number of syncytia (multinucleated giant cell) in each well of 96-well plates under an inverted microscope (100X). The inhibitory percentage of syncytia formation was calculated by the percentage of syncytia number in a treated sample compared to that in infected control. 50% effective concentration (EC50) was calculated.
Formula
According to the method described by Reed and Munch, 50% cytotoxic concentration (CC50) and 50% effective concentration (EC50) was determined from dose-response curve. Therapeutic index (TI) of anti-HIV activity is CC50/EC50.
1. Cell viability (% of control)=(ODtest-Odblk)/(Odctrl-Odblk) × 100
2. CPE inhibition (%)=(1-CPEtest /CPEctrl) × 100
Antibacterial and antifungal activity
Minimum inhibitory concentrations (MICs) were generally used to find out and to estimate the activity of various synthesized compounds against numerous pathogenic and nonpathogenic bacteria as well as fungi. Antibiotics like Ciprofloxacin and Norfloxacin were used as the standard in case of antibacterial activity and fluconazole was used as standard in case of antifungal activity of the synthesized compounds (5a-5e). Most of the tested compound was found active against Gram-positive and Gram-negative bacteria. Gram positive and Gram negative bacteria were estimated as minimum inhibitory concentration values (MICs) against several synthesized compounds.
All the synthesized compounds (5a-5e) were evaluated for their antibacterial activity against Gram-positive bacteria such as Staphylococcus aureus (NCIM2122), Bacillus subtilis (MTCC121) and Gram-negative bacteria: Escherichia coli (MTCC 118), Pseudomonas aeruginosa (MTCC 647), Salmonella Typhi (NCIM 2501) and Klebsiella pneumonia (MTCC 3384) through two fold serial dilution method. All the synthesized compounds (5a-5e) were evaluated for their antifungal activity against Candida albicans (MTCC 227) and Aspergillus niger (NCIM 1056) and fluconazole was used as the standard.
A culture of pure and individual microorganism was grown in Mueller Hinton broth. The antimicrobial compounds were diluted several times in 1: 1 ratio, through a sterile diluent (usually Mueller Hinton broth). All test compounds were dissolved in approximately 10% Dimethyl sulphoxide (DMSO) in order to prepare the stock solution of 2000 g/ml. These solutions were serially diluted to prepare a further concentration of 100, 50, 25, 12.5, 6.25, and 3.125 μg/ml. The Mueller- Hinton Broth nutrient media was used for bacteria while Sabouraud Dextrose Broth media used for fungus. The cellular density of microbes was adjusted through the sterile water of a 0.5 McFarland standard. A final concentration was ~107 CFU/mL for bacteria and ~106 CFU/mL for fungus respectively. Two fold diluted samples were added to the Microbial inoculums. The diluted test tubes were incubated for 18-24 h at 37°C ± 1°C in case bacteria and for 2-5 days at 25°C ± 1°C in case of fungus. The highest dilution of the test compounds which can completely inhibit the growth of microorganism was considered as the MIC (Minimum Inhibitory Concentration) and value of the test compounds were expressed in μg/ml.
Anthelmintic activity
The measure was executed on Indian earthworm, Pheretima posthuma because of its anatomical and physiological closeness with intestinal roundworm parasite of people. An anthelmintic measure was executed according to the technique is given in the literature. All the earthworms were collected from Divyayan Krishi Vigyan Kendra, Jharkhand, India. The earthworm, Pheretima posthuma washed with typical saline solution (0.5%) for around 30 sec to evacuate all fecal issue were utilized for an anthelmintic investigation. Earthworms of 2-4 cm long were utilized as a part of this analysis and were separated into three gatherings of six each. Each one of the mixes was broken down in least amount of 2% v/v Tween 80. Before beginning the tests, every one of the mixes and standard medication arrangements were naturally arranged. All the combined mixes were subjected to contemplate anthelmintic activity against worms at 5 mg/mL, 10 mg/mL, and 20 mg/mL concentrations. The paralyzing and death times were noted and their mean was computed for triplicate sets. Death time was recorded by putting earthworms in warm water and watched for stimulated movement, if the worm was alive. Albendazole was utilized as a standard drug at a measurement of 20 mg/ml.
Statistical analysis
Results were expressed as mean ± S.D. Significance determined by one-way analysis of variance (ANOVA) followed by Dunnett’s test with level of significance at P<0.01 and P<0.05.
Results and Discussion
Chemistry
A group of five imidazole analogs (5a-5e) was blended after the response is appeared in Scheme 1. The synthesis of the title compounds was completed in 4 steps.
To begin with ammonium phenyl carbamodithioate (2) was synthesized. Ammonium phenyl carbamodithioate was broken down in 10 ml of methanol with cooling in an ice bath. Methyl iodide was added to an mixture and stirred vigorously at temperature 0ºC-4ºC for 3 h. An intermediate as methyl phenyl carbamodithioate (3) was disintegrated in 15 ml of methanol at 60ºC. 85% hydrazine hydrate was added to the reaction mixture and mixed consistently for 2 h a phenyl thiosemicarbazide was shaped. (4) To a mixture of para substituted phenacyl bromide in glacial acetic acid, Potassium thiocyanate was added with continous mixing at the temperature of 60ºC. The reaction mixture was then mixed for 30 min at same temperature. An intermediate as phenyl thio semicarbazide was added to reaction mixture and mixing was proceeded at 60ºC for 60 min. The reaction mixture then refluxed for 6 hours. In the wake of refluxing was finished, 30 ml of water was added to the reaction mixture which yielded a sticky mass. It was washed with ether and product was accelerated out as an amorphous powder and recrystallized from an ethanol-water mixture.
A final product of para substituted phenyl-2-mercapto-1-H-imidazol-1-yl-3-phenyl thiourea derivatives have been synthesized in four steps (5a-5e).
The IR (KBr) spectrum of all the synthesized compounds showed very similar frequencies indicating that all the synthesized compounds have very similarities with in the structures for the title compounds. The IR spectra of the synthesized compounds contained the thiocarbonyl stretching (C=S) band 1731-1600 cm-1. Similarly, N-H stretching band was seen between 3200-3000 cm-1. The C-H band in IR spectra of all the compounds appeared at 2100-2000 cm-1 and C-N absorption range from 1300-1200 cm-1.
The 1H-NMR spectrum of the title compounds (5a-5e) was recorded in Deuterated chloroform (CDCl3) solution and having a predictable resonance signal in form of chemical shifts and their integration values. 1H-NMR spectrum of the title compounds (5a-5e) showed a broad singlet of 1 proton assigned to SH proton at δ 1.20-2.50. Depending on the nature of the substituent's and substitution pattern on the N -phenyl ring, the aromatic protons of certain compounds (5a-5e) were observed in distinct chemical shifts with specific splitting patterns as doublets, triplets, or multiplets. The integration unit is more than one proton due to the close chemical shifts ranging from δ 5.530-8.020. In the aliphatic region, a broad singlet of 3 protons assigned to the methyl or methoxyl proton of -CH3 or OCH3 ranges δ (ppm)=2.20-2.86 was observed for the compounds. The structural confirmations of these compounds were determined by using Mass spectroscopy (ESI-MS).
Anti-HIV activity
All the compounds were screened for their in vitro Anti-HIV activity. The results are given in Table 2.
| Compounds | Experiment | Method | CC50 (µM) | EC50 (µM) | Therapeutic Index (TI) |
|---|---|---|---|---|---|
| 5a | Cytotoxicity assay | MTT | 87.2 | - | 17.5 |
| Inhibition of syncytium formation | CPE | - | 4.98 | ||
| 5b | Cytotoxicity Assay | MTT | 64.35 | - | 19.9 |
| Inhibition of syncytium formation | CPE | - | 3.23 | ||
| 5c | Cytotoxicity Assay | MTT | 36.4 | - | 3.4 |
| Inhibition of syncytium formation | CPE | - | 10.72 | ||
| 5d | Cytotoxicity Assay | MTT | 31.01 | - | 9.1 |
| Inhibition of syncytium formation | CPE | - | 3.4 | ||
| 5e | Cytotoxicity Assay | MTT | 71.36 | - | 8.6 |
| Inhibition of syncytium formation | CPE | - | 8.28 | ||
| AZT | Cytotoxicity Assay | MTT | 1291 | - | 514342.6 |
| Inhibition of syncytium formation | CPE | - | 0.00251 |
Table 2: The summary of cytotoxicity and Anti-HIV-1 activities of compound
Among all the series of compounds 5a-5e; Compounds 5a and 5b showed a significant degree of anti-HIV activity compared to zidovudine (AZT) because of electron withdrawing group as substituent showed therapeutic index was 17.5 and 19.9 while compound 5d and 5e shows moderate activity and shows therapeutic index 9.1 and 8.6 respectively. Compound 5c showed less significant activity against a standard. while cytotoxicity concentration (CC50) in μM by MTT method of compound 5a, 5b, 5c, 5d, and 5e were found to be 87.2, 64.35, 36.4, 31.01 and 71.36 The effective concentration of synthesized compounds 5a-5e, compound 5a, 5b, 5c, 5d and 5e were found to be very effective and that were 4.98, 3.23, 10.72, 3.40 and 8.28 respectively which were less than standard zidovudine (AZT) that is (EC50=0.00251 μg/ml). As a control, AZT has the best anti-HIV activity (EC50=0.00251 μg/ml) in vitro, and the CC50 of is 1291.00 μg/ml, its therapeutic index is 514342.6 (Figure 5).
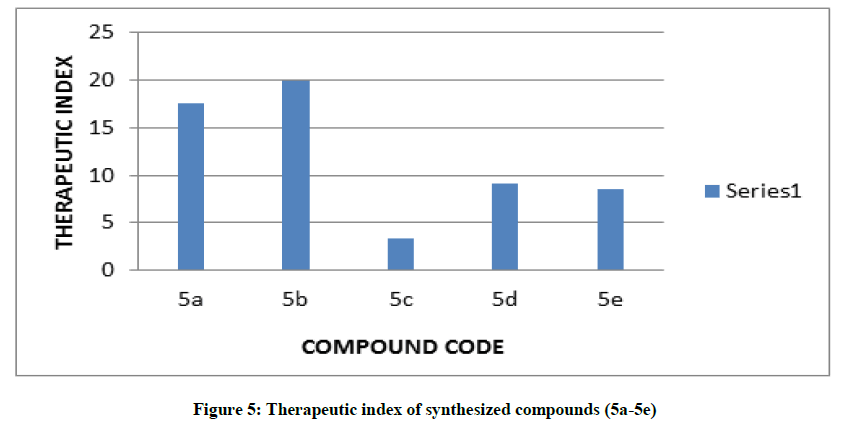
Figure 5: Therapeutic index of synthesized compounds (5a-5e)
Microbial study
Antibacterial activity
All the synthesized compounds (5a-5e) were evaluated for in-vitro antibacterial activity against two strain of Gram-positive bacteria: B. subtilis (MTCC 121), S. aureus (NCIM 2122), and four strain of Gram-negative bacteria: E. coli (MTCC 118), P. aeruginosa (MTCC 647), S. typhi (NCIM 2501), K. pneumoniae (MTCC 3384) and two strain of fungus C. albicans (MTCC 227), A. niger (NCIM 1056), by using twofold serial dilution technique and the results are summarized in Tables 3 and 4. Ciprofloxacin was used as the standard for antibacterial activity and fluconazole was used as the standard for antifungal activity.
| Microorganisms | ||||||
|---|---|---|---|---|---|---|
| Gram (+) bacteria | Gram (-) bacteria | |||||
| Compounds | Staphylococcus aureus | Bacillus subtilis | Escherichia coli | Pseudomonas aeruginosa | Salmonella typhi | Klebsiella pneumoniae |
| 5a | 6.25 | 3.125 | 6.25 | 6.25 | 3.125 | 3.125 |
| 5b | 6.25 | 3.125 | 6.25 | 3.125 | 6.25 | 6.25 |
| 5c | 12.5 | 12.5 | 25 | 12.5 | 25 | 12.5 |
| 5d | 6.25 | 12.5 | 12.5 | 25 | 25 | 12.5 |
| 5e | 25 | 12.5 | 25 | 12.5 | 12.5 | 6.25 |
| Ciprofloxacin | ≤ 1 | ≤ 1 | ≤ 1 | ≤ 1 | ≤ 1 | ≤ 1 |
| Norfloxacin | ≤ 1 | ≤ 1 | ≤ 1 | ≤ 1 | ≤ 1 | ≤ 1 |
Table 3: Minimum Inhibitory Concentration (MIC) of Test Compounds 5a to 5e against Staphylococcus aureus, Bacillus subtilis, Salmonella Typhi, Escherichia coli, Pseudomonas aeruginosa and Klebsiella pneumonia
| Micro-organisms (fungal strain) | ||
|---|---|---|
| Compounds | Candida Albicans | Asperigillus Niger |
| 5a | 3.125 | 6.25 |
| 5b | 6.25 | 6.25 |
| 5c | 12.5 | 12.5 |
| 5d | 12.5 | 6.25 |
| 5e | 12.5 | 12.5 |
| Fluconazole | 12.5 | 12.5 |
Table 4: Minimum Inhibitory Concentration (MIC) of Test Compounds 5a to 5e against Candida albicans and Asperigillus niger
The antimicrobial drugs available in the market have numerous drawbacks such as toxicity, narrow spectrum of activity and some of the antibiotics also exhibit drug-drug interactions. And due to high chances of infections in immune compromised patients, demands for some good and effective antimicrobial agents with a broad spectrum of activity and good pharmacokinetic properties have increased. Most of the compounds exhibited mild to moderate antibacterial activity against various Gram positive and Gram - bacterial strains. The antibacterial data indicated that all compounds ranges from (5a-5e) illustrated appreciable antibacterial activity against various strains but lower than that of standard ciprofloxacin. Antibacterial activity of compounds (5a-5e) (MIC in μg/ml) is summarized in Table 3.
Antifungal activity
All the synthesized compounds (5a-5e) were evaluated for their antifungal activity against C. albicans (MTCC 227) and A. niger (NCIM 1056). Most of the compounds exhibited very high antifungal activity against C. albicans (MTCC 227) and A. niger (NCIM 1056). Compounds 5a, 5b, and 5e showed potent antifungal activity against C. albicans (MTCC 227) and A. niger (NCIM 1056) comparable and even higher than that of standard fluconazole. Antifungal activity of compounds (5a-5e) (MIC in μg/ml) is shown in Table 4.
Anthelmintic activity
The in vitro anthelmintic activity of test compounds (5a-5e) was assessed in comparison to standard drug albendazole against Pheretima Posthuma. The doses of the test compounds used were 5 mg/ml, 10 mg/ml and 20 mg/ml. The mean paralysis time and lethal time were calculated for each test compounds and standard drug. It is evident that the all the test compounds (5a-5e) exhibited anthelmintic activity in a concentration-dependent manner giving the shortest time of paralysis and death at 20 mg/ml. It was observed that the time taken for paralysis and death was least for test compounds (5c), and (5d) at all concentrations. SAR studies showed that presence of para-substituted halogens and the para-substituted methyl group on the phenyl ring played a significant role in an increase in anthelmintic activities. Anthelmintic activity of test compound (5a-5e) against Pheretima Posthuma is shown in Table 5.
| Test | Time taken for paralysis (P) and death (D) (Mean ± SD) | |||||
|---|---|---|---|---|---|---|
| Compound | Paralysis Time (min) Lethal Time (min) | |||||
| 5 mg | 10 mg | 20 mg | 5 mg | 10 mg | 20 mg | |
| 5a | 21.7 ± 0.45 | 14.9 ± 0.70 | 6.10 ± 0.43 | 28.70 ± 0.43 | 20.60 ± 0.62 | 11.90 ± 0.62 |
| 5b | 20.87 ± 0.31 | 18.90 ± 0.41 | 9.26 ± 0.18 | 32.60 ± 0.36 | 25.00 ± 0.35 | 11.20 ± 0.21 |
| 5c | 10.5 ± 0.51 | 8.66 ± 0.61 | 6.34 ± 0.10 | 14.93 ± 0.26 | 12.86 ± 0.68 | 9.96 ± 0.49 |
| 5d | 17.66 ± 0.47 | 13.63 ± 0.47 | 8.67 ± 0.66 | 23.73 ± 0.47 | 18.00 ± 0.55 | 10.26 ± 0.60 |
| 5e | 22.43 ± 0.22 | 18.13 ± 0.51 | 8.63 ± 0.55 | 26.33 ± 0.58 | 23.90 ± 0.65 | 15.34 ± 0.15 |
| Albendazole | 5.43 ± 0.34 | 7.52 ± 0.36 | ||||
Table 5: Anthelmintic activity of test compounds 5a to 5e against Pheretima posthuma
All determinations were done in triplicate and results are expressed as mean ± SD. N=6, p value was calculated by comparing with control by one-way ANOVA. P<0.05, P<0.01 and P<0.001, significantly different when compared with reference compound, Albendazole.
Determination of minimal inhibitory concentrations (MICs)
Minimum inhibitory concentration (MIC) is the lowest concentration of the drug which inhibits the growth of the bacteria and fungi. The data on the antimicrobial activity of the compounds and the standard drugs as MIC values are given in Table 3 and Table 4.
Conclusion
A group of five imidazole analogs (5a-5e) have been synthesized and their chemical structures have been identified by IR, NMR (Appendix Figures 1-10) and Mass spectroscopy. Synthesized compounds (5a-5e) were evaluated for their anti-HIV, antimicrobial (antibacterial and antifungal) and anthelmintic activities followed by two fold serial dilution method. From anti HIV activity it must be presumed that electron withdrawing group like chloro and bromo at para position gives preferable action over electron donating group like methyl and methoxy while if there should arise an occurrence of antibacterial and antifungal activities, it must be reasoned that fuse of electron donating/electron withdrawing back gatherings in the in both the aryl moieties joined to the aryl/heteroaryl pharmacophore was a required component for high antibacterial and antifungal exercises. Present studies encourage us to consider this a newer molecular skeleton of para substituted phenyl-2-mercapto-1-H-imidazol-1-yl-3-phenyl thiourea which may be identified as a potential scaffold for the development of wide-spectrum chemotherapeutic agents. Anthelmintic activity observed that the time taken for paralysis and death of earthworm was better for methyl and methoxyl substituent.
Acknowledgement
The authors are thankful to Dr. (Prof.) Yong-Tang Zheng and his team, Laboratory of Molecular Immunopharmacology, Kunming Institute of Zoology, Chinese Academy of Science, for Anti-HIV activity as MTT colorimetric assay and syncytia formation assay. The authors also thank to Dr. Reddy’s Institute of Life sciences, Hyderabad as well as BITS Pilani for spectral characterization. One of the authors (ROHIT SINGH) gratefully acknowledges the University Grants Commission-Major Research Project (UGC-MRP letter No. F. No. 42-690/2013(SR) for the award of fellowship during the work.
Conflict of Interest
The authors confirm that content of this article has no conflict of interest.
References
- http://www.who.int/gho/hiv/en/
- K. Das, E. Arnold, Current opinion in virology., 2013, 2, 119-28.
- L.H. Jones, T. Dupont, C.E. Mowbray, S.D. Newman, Organic letters., 2006, 8, 1725-1727.
- E.D. Clercq, Chemistry & biodiversity., 2004, 1, 44-64.
- R. Singh, S. Ganguly, Anti infective agents.,2018, 16, (Accepted and in press).
- J. Ren, C. Nicolas, L. E. Bird, T. Fujiwara, H. Sugimoto, D. Stuart, Journal of Biological Chemistry. 2000, 275, 14316-14320.
- I.M. Lagoja, C. Pannecouque, Z. Debyser, J. Balzarini, A.V. Aerschot, M. Witvtouw, P.J. Herdewijn, J. Med. Chem., 2003, 46, 1546-1553.
- K. Maeda, T. Osata, H.A. Umezawa, J. Antibiot., 1953, 6, 182-184.
- W.J. Watkins, T.E. Renau, Burger’s Medicinal Chemistry & Drug Discovery, 6th (Edn.), 2003, 5, 893-899.
- R. Griffith, T. Tracy, Foye’s principles of Medicinal Chemistry, 5th (Edn.), 2002, 894-899.
- E.F. Elslager, P.E. Thompson, Ann. Rev. Pharmacol., 1962, 2, 193-214.
- E.F. Elslager, Ann. Rept. Med. Chem., 1966, 1, 136-149.
- E.F. Elslager, Human Antiparasitic Agents Ann. Rept. Med. Chem., 1967, 2, 131-146.
- P. Bousquet, J. Feldman, J. Schwartz, J. Eur. J. Pharmacol., 1984, 230, 232-236.
- E. Urech, A. Marxer, K. Miescher, Helv. Chim. Acta., 1950, 33, 1386-1407.
- R.R. Lee, B. Justin, C. Kelly, M. Gordon, S. Alan, A.W. Gavin, Bioorg. Med. Chem. Lett., 2000.
- Y.H. Sung, W.K. Kyu, K.R. Chung, J.K. Soo, H.C. Kwang, Bioorg. Med. Chem., 2008, 16, 644-649.
- H.L. Nan, W. Le, W. Xilu, T.W. Gary, C. Jerry, G. Wen, Z. Haiying, H.R. Saul, L.S. Hing, Bioorg. Med. Chem. Lett., 2004, 14, 5057-5062.
- S. Kristina, K. Marijeta, E. Katja, Bioorg. Med. Chem., 2007, 15, 4419-4426.
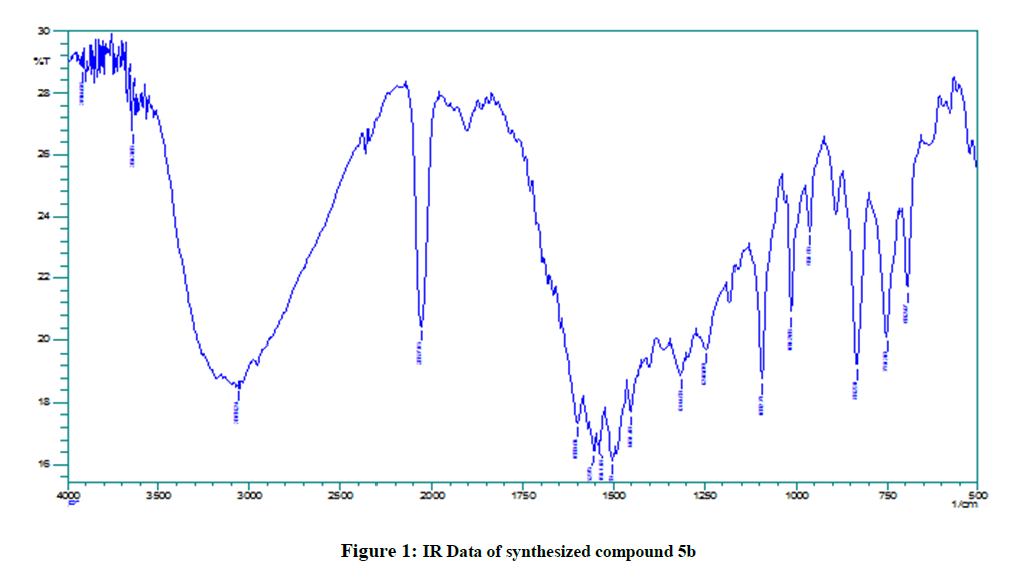
Figure 1: IR Data of synthesized compound 5b
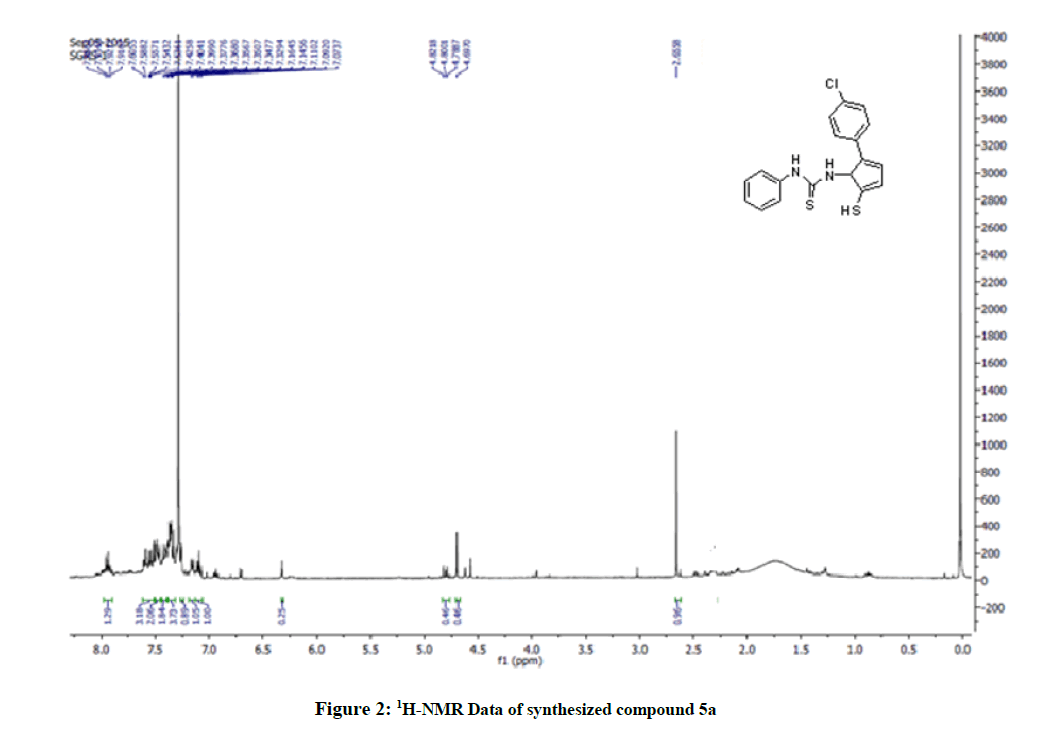
Figure 2: 1H-NMR Data of synthesized compound 5a
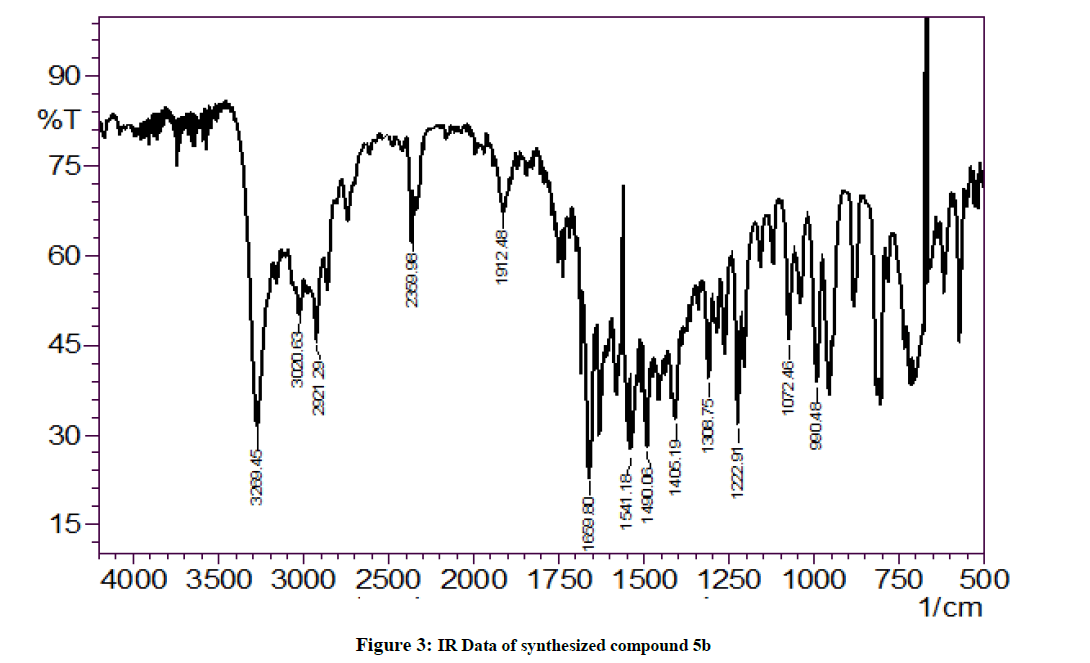
Figure 3: IR Data of synthesized compound 5b
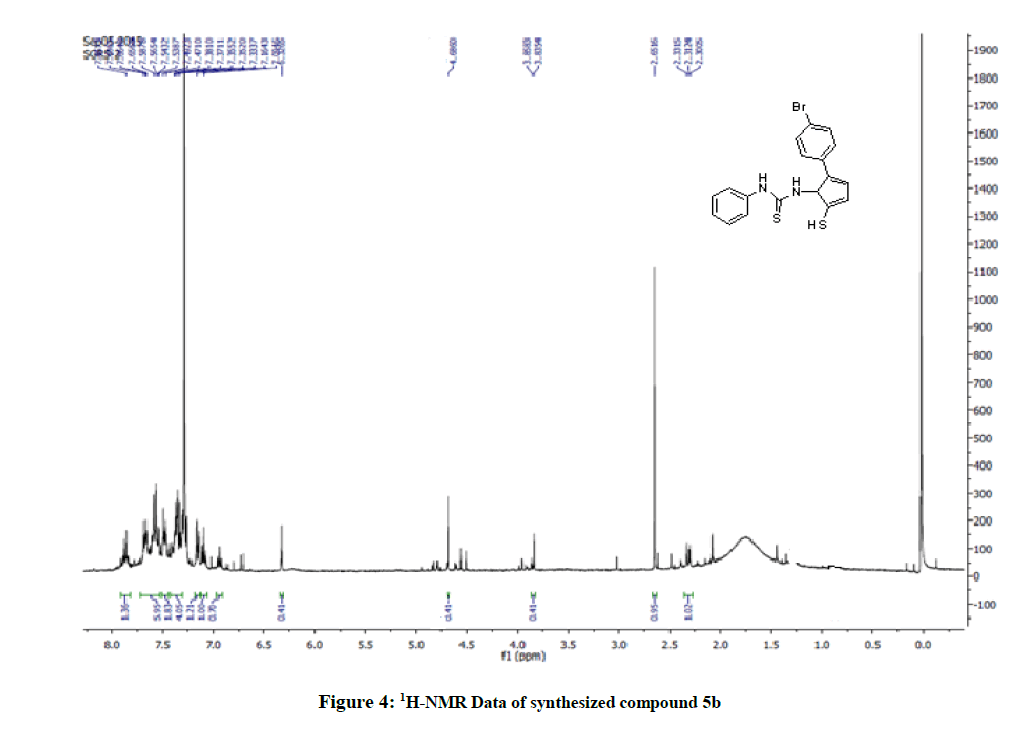
Figure 4: 1H-NMR Data of synthesized compound 5b
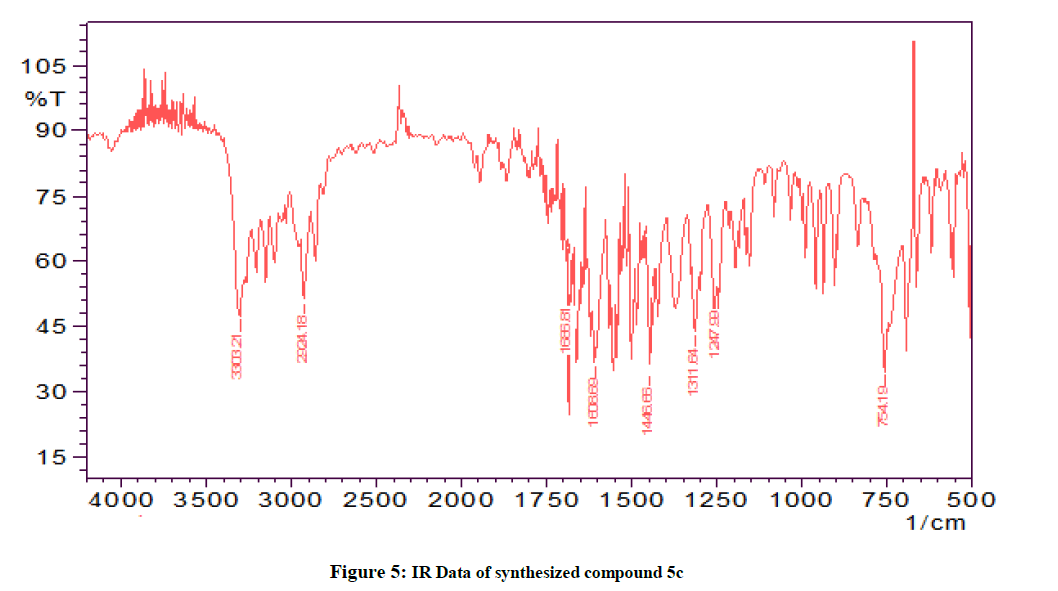
Figure 5: IR Data of synthesized compound 5c
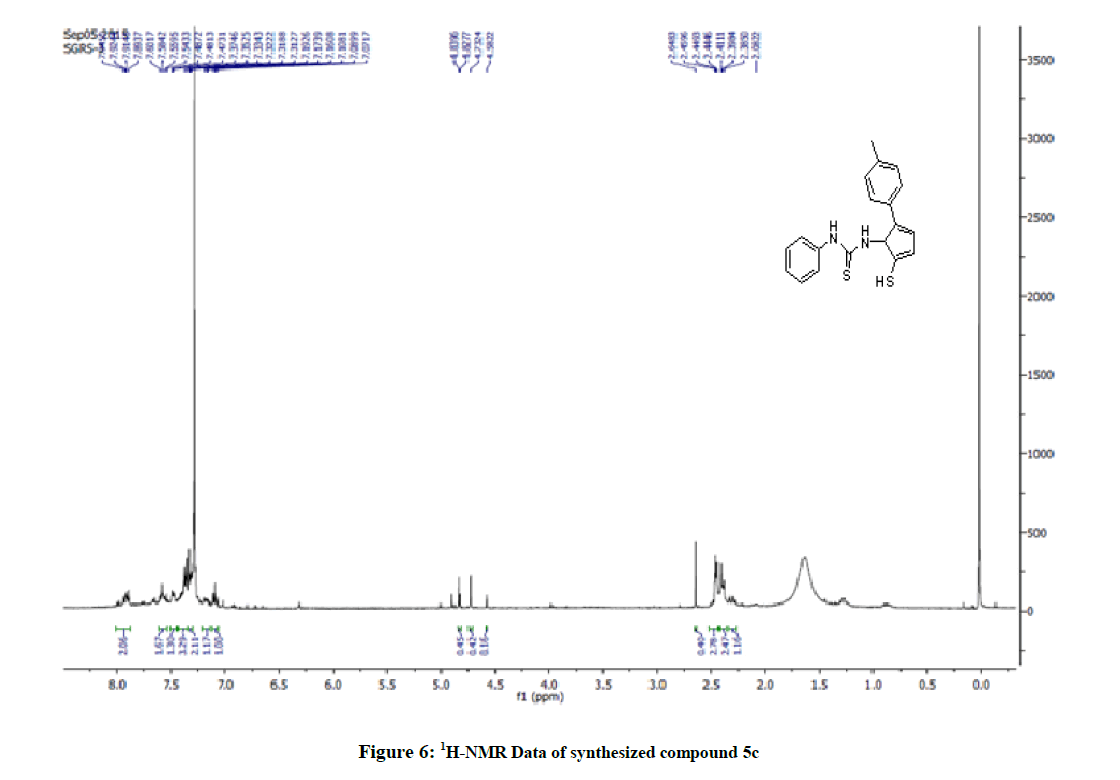
Figure 6: 1H-NMR Data of synthesized compound 5c
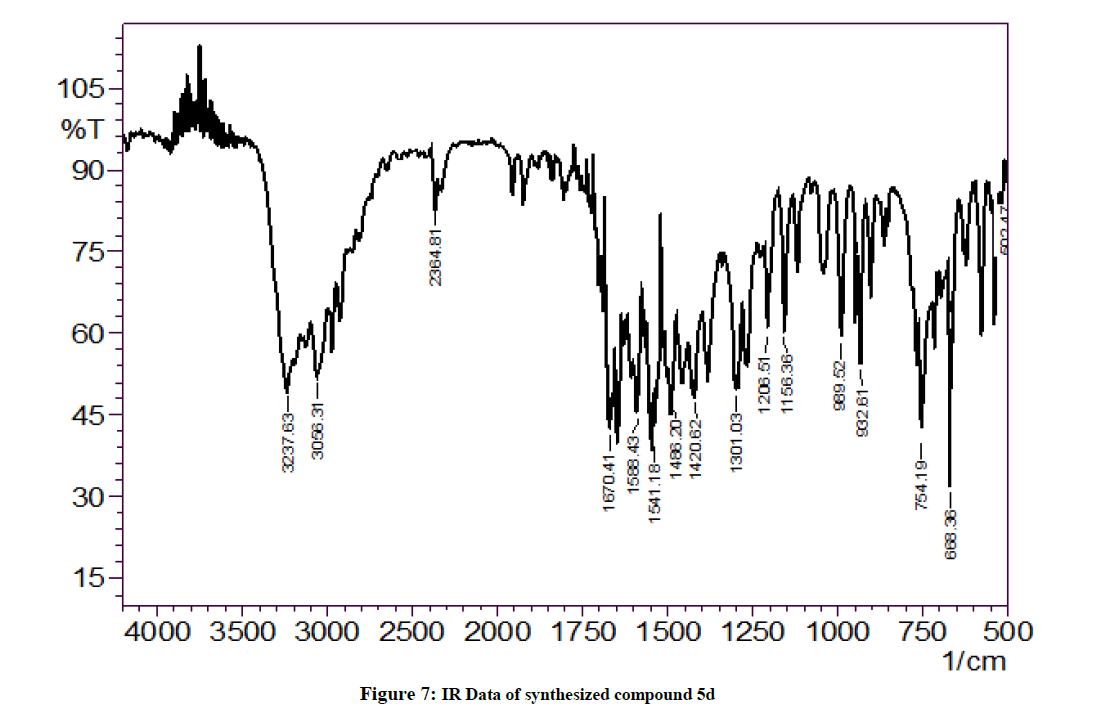
Figure 7: IR Data of synthesized compound 5d
Figure 8: 1H-NMR Data of synthesized compound 5d
Figure 9: IR Data of synthesized compound 5e
Figure 10: 1H-NMR Data of synthesized compound 5e



