Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 6
Synthesis, In Vitro Anticancer Evaluation, and In Silico Studies of New Thiazolo[3,2-A]Pyrimidin-5-One Derivatives
Ahmed R. Ali, Eman R. El-Bendary, Mariam A. Ghaly* and Ihsan A. Shehata
Department of Medicinal Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
- *Corresponding Author:
- Mariam A. Ghaly
Department of Medicinal Chemistry
Faculty of Pharmacy
Mansoura University
Mansoura 35516, Egypt
Abstract
The present study involves the development of certain thiazolo[3,2-a]pyrimidin-5-ones linked through an ethylene bridge to various amines. The newly synthesized compounds 4-6(a-c) were subjected to in vitro anticancer evaluation using NCI antitumor screening. The target compounds showed observed activity against Renal UO-31 cancer cell line with cell growth promotion 52.72%-64.52%. Assessment of toxicities, druglikeness, and drug score profiles are reported. Some of the synthesized compounds showed good docking scores with potential anticancer targets. In vitro anticancer evaluation, together with in silico studies, revealed that compounds 5c, 4a, and 4b could be considered as promising leads for further development of more potent anticancer agents.
Keywords
Aminothiazole, Thiazolopyrimidine, Anticancer evaluation.
Introduction
Fused thiazoles are an important class of compounds which have attracted much attention to make use of their remarkable biological and pharmacological properties. Several publications have pointed to the antitumor activity of fused thiazole compounds e. g. thiazolo[5,4-c]pyridin- 4(5H)-one [1], and thiazolo[3,2-a]pyrimidine derivatives [2-5].
Based on these findings, and in continuation to our efforts to synthesize biologically active compounds against cancer [6,7], we became interested in the evaluation of a series of thiazolo[3,2-a]pyrimidin-5-one derivatives with various substituents at the 3-, 6- and 7-positions, aiming at identifying potent anticancer agents.
Several reports indicated that potent kinase inhibitors of epidermal growth factor receptor, for example gefitinib for non-small-cell lung cancer [8], mitogen-activated protein [9], and proto-oncogene tyrosine-protein [10] encompassed diversity of heterocycles bearing alkyl bridge with a morpholine moiety. A morpholine ring is often introduced to enhance water solubility. The group is attached through alkyl chain in order to protrude from the binding site and be exposed to the surrounding aqueous environment [11].
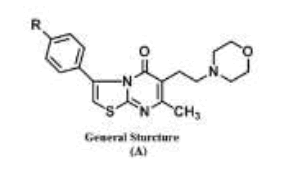
On the basis of the preceding information, a part of the research undertaken here involved the combination of the morpholinoalkyl moiety with the thiazolo[3,2-a]pyrimidin-5-one series in a single molecular frame of the general structure (A) with the hope of finding interesting antitumor activity.
Moreover, it is well documented that aryl/heteroaryl sulfonamides, where the nitrogen of -SO2NH2 group is either free or substituted, exhibited substantial antitumor activity in vitro and/or in vivo [12,13]. The discovery of E-7010 [14] and vemurafenib (PLX4032) [15], fused heterocyclic compounds incorporating sulfonamide moiety, emphasized the role of sulfonamides as an important class of anticancer agents which interact with a wide range of different cellular targets. In addition, series of novel compounds containing benzenesulfonamide moiety and incorporating benzoquinones [16], quinazolin-2-ones [17] or coumarins [18] have revealed promising anticancer activities.
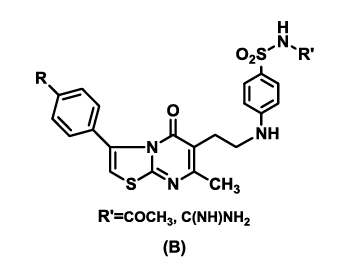
In view of the preceding information, it was envisaged to construct a system which combines both thiazolo[3,2-a]pyrimidin-5-ones and sulfonamides in a single molecular frame (B), in order to explore the additive effects towards their anticancer activities.
Results and Discussion
Chemistry
A general approach to synthesize the designed compounds 4-6(a-c) is shown in Scheme 1. 2-Amino-4-arylthiazoles 2a-c were prepared utilizing either phenacyl chloride or bromide according to a reported procedure [19] which is considered to be an easy, rapid and purification-free procedure. Thiourea was allowed to react with phenacyl halide at room temperature for 2-3 minutes to yield the corresponding arylthiazole. The reaction of 2a-c with α-acetyl-γ-butyrolactone in phosphorus oxychloride afforded 6-(2-chloroethyl)-7-methyl-3-(un)substituted phenyl-5H-thiazolo[ 3,2-a]pyrimidin-5-ones 3a–c without isolating the intermediates [20,21] in quantitative yields.
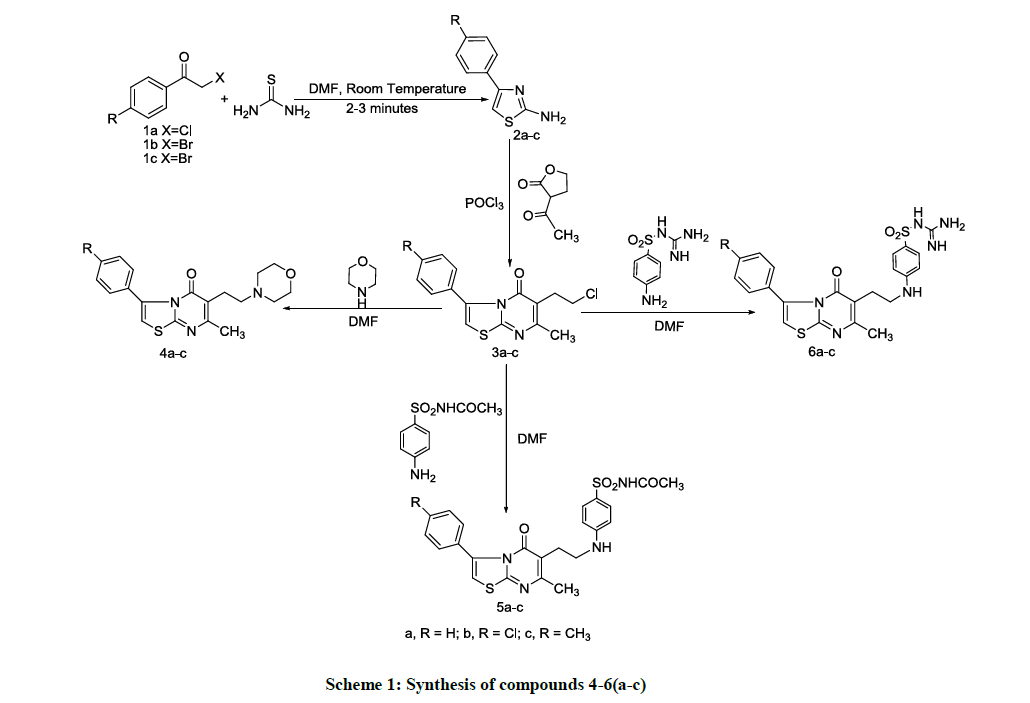
Scheme 1: Synthesis of compounds 4-6(a-c)
Heating compounds 3a-c with morpholine, sulfacetamide, and sulfaguanidine in dry DMF in the presence of triethylamine gave the corresponding target compounds 4-6(a-c) in moderate yields.
The structures of the synthesized compounds 4-6(a-c) were confirmed by microanalyses and spectral data (IR, 1H-NMR, 13C-NMR and EI-MS) which showed full agreement with their structures. In the 1H-NMR spectra of compounds 4a-c, the triplet signals of the morpholine ring protons resonated at the expected regions integrating for eight protons. In the 13C-NMR spectra of compound 4a, new bands appeared at 53.16 and 66.15 ppm, attributed to ((CH2)2N-morpholine) and ((CH2)2O-morpholine), respectively. For compounds 5a-c, the aromatic protons (-NH-C6H4- SO2NH-) in the 1H-NMR spectra and the 13C signals of COCH3 and COCH3 in compound 5a were observed at the expected regions. The 13C signal for guanidine moiety in compound 6a was observed at 159.23 ppm. The mass spectral data of the synthesized compounds 4-6(a-c) displayed molecular ion peaks which confirmed their molecular weights.
Biological evaluation
In vitro anticancer screening
The target compounds 4-6(a-c) were submitted to the National Cancer Institute (NCI) [22], Bethesda, Maryland, USA, under the Developmental Therapeutic Program (DTP). The operation of this screen utilizes 60 different human tumor cell lines, representing leukemia, melanoma and cancers of the lung, colon, brain, ovary, breast, prostate, and kidney. Structures are generally selected for screening based on their ability to add diversity to the NCI small molecule compound collection. Compounds with drug-like properties based on computer-aided design are to be prioritized in the NCI screening service. All compounds submitted to the NCI 60 cell screen were tested initially at a single high dose (10-5 M) in the full NCI 60 cell panel. The compounds were added at a single concentration (10-5 M) and the culture was incubated for 48 h. End point determinations were made with a protein binding dye, Sulforhodamine B [23-25].
The mean percentage growth percentages and the growth percentage with the most sensitive cell lines of all of the tested compounds over the full panel of cell lines are illustrated in Table 1.
| Comp. NO. | NSC code | Mean Percentage Growth | Range of Growth | Leukemia SR | Non-Small Cell Lung Cancer HOP-92 | CNS Cancer SNB-75 | Renal Cancer UO-31 | Prostate Cancer PC-3 | Breast Cancer MDA-MB-231/ATCC | Breast Cancer T-47D |
|---|---|---|---|---|---|---|---|---|---|---|
| 4a | 768162 | 96.24 | 77.6 | 70.28 | 84.4 | 71.77 | 58.46 | 81.37 | 79.26 | 84.65 |
| 4b | 768163 | 96.91 | 84.42 | 81.39 | 81.6 | 84.59 | 57.53 | 87.5 | 78.32 | 76.61 |
| 4c | 768178 | 96.88 | 77.94 | 81.78 | 70.44 | 73.56 | 64.52 | 79.48 | 79.66 | 88.13 |
| 5a | 768164 | 97.82 | 73.08 | 75.05 | 82.43 | 82.31 | 55.24 | 84.74 | 81.15 | 78.49 |
| 5b | 768165 | 99.22 | 78.84 | 91.86 | 80.43 | 87.92 | 57.26 | 86.69 | 83.53 | 82.39 |
| 5c | 768186 | 95.98 | 78.77 | 89.43 | 62.58 | 75.52 | 62.94 | 73.44 | 73.92 | 84.24 |
| 6a | 768166 | 96.18 | 79.86 | 81.71 | 79.02 | 87.97 | 52.72 | 83.75 | 79.61 | 89.62 |
| 6b | 768167 | 95.54 | 71.13 | 92 | 66.69 | 80.01 | 53.92 | 80.27 | 79.32 | 84.05 |
| 6c | 768187 | 99.94 | 90.94 | 95.05 | 70.69 | 73.13 | 61.63 | 85.53 | 83.98 | 85.76 |
Table 1: Mean percentage growth and screening data of the final compounds with the most sensitive cell lines represented as percent cell growth
In light of the NCI results, the following could be considered:
Regarding the sensitivity against individual cell lines in Table 1, all target compounds 4-6(a-c) showed observed low cell growth promotion against Renal UO-31 cancer cell line with cell growth promotion varying from 52.72% to 64.52%.
By comparing the results from different series, it was found that the introduction of sulfacetamide in compounds 5a-c or sulfaguanidine in compounds 6a-c instead of morpholine moiety in compounds 4a-c proved to enhance the potency towards Renal UO-31 cancer cell line and reduce potency towards Leukemia SR cancer cell line.
It is worth mentioning that compounds 4-6(a) exhibited increased potency towards Leukemia SR cancer cell line and reduced the potency towards Non-Small Cell Lung HOP-92 cancer.
Assessment of toxicities, druglikeness, and drugscore profiles
Osiris program [26] was used for prediction of the overall toxicity of the designed derivatives as the prediction process relies on a predetermined set of structural fragments that give rise to toxicity alerts in case they are encountered in the structure. All target compounds 4-6(a-c) showed low in silico possible toxicity risks as shown in Table 2. Osiris program was also used for calculating the fragment-based druglikeness of the designed compounds. A positive value states that the designed molecule contains fragments which are frequently present in commercial drugs.
| Comp. No. | Toxicity Risks(Mutagenicity, Tumorigenicity, Irritancy, Reproductive Effects) | Druglikeness | Drug Score |
|---|---|---|---|
| 4a | -a) | 4.9 | 0.84b) |
| 4b | -a) | 5.48 | 0.75 |
| 4c | -a) | 3.65 | 0.80b) |
| 5a | -a) | 7.93 | 0.59 |
| 5b | -a) | 8.42b) | 0.47 |
| 5c | -a) | 6.64 | 0.54 |
| 6a | -a) | 7.47 | 0.66 |
| 6b | -a) | 7.95b) | 0.55 |
| 6c | -a) | 6.16 | 0.61 |
a) No indication for toxic effects; b) Underlined values represent the highest results in each parameter
Table 2: Toxicity risks, druglikeness, and drug scores of the tested compounds
The drug score combines druglikeness, cLogP, LogS, molecular weight and toxicity risks in one handy value that may be used to judge the compound's overall potential to qualify for a drug. A value of 0.5 or more makes this compound a promising lead for future development of safe and efficient drug. Predictions of potential toxicity, druglikeness, and drug score for the studied compounds are given in Table 2. Almost all of the synthesized compounds possess good values of druglikeness and drug score. Compound 4a, with the highest drug score value, showed the highest potency over Leukemia SR and CNS SNB-75 cancer cell lines and considerable potency against Renal UO-31 and Breast MDA-MB- 231/ATCC cancer cell lines.
Target Fishing
An attempt was made to investigate the potential targets involved in observed inhibition displayed by the synthesized compounds against NCI 60 cell panel. PharmMapper server is a freely accessed web server designed to identify potential target candidates for the given small molecules using reverse pharmacophore mapping approach [27].
PharmMapper is available at http://59.78.96.61/pharmmapper. The newly synthesized compounds 4-6(a-c) were uploaded in Tripos Mol2 or MDL SDF format. PharmMapper adopts semi-rigid pharmacophore mapping protocol. As a result, multiple conformations of the query molecule are required prior to mapping which can be achieved by online service provided by the server. PharmMapper demonstrated a variety of putative targets that might exhibit considerable binding affinity to the synthesized compounds. Six targets, involved in cancer therapy, are common between the synthesized compounds. These targets might explain the observed antiproliferative activity. The fit scores of the synthesized compounds with the top targets were illustrated in Table 3.
| Fit Score With Different Enzymes (PDB-Id) | ||||||
|---|---|---|---|---|---|---|
| Comp. No. | Cathepsin K (1TU6) | Vitamin D3 receptor (1DB1) | Dual specificity mitogen-activated protein kinase kinase 1 (1S9J) | Proto-oncogene tyrosine-protein kinase Src (1Y57) | Epidermal growth factor receptor (1XKK) | Leukotriene A4 hydrolase (1GW6) |
| 4a | 3.75 | 3.15 | 3.15 | 3.31 | 2.98 | 3.62 |
| 4b | -a) | 3.61 | 3.68 | 3.31 | 3.55b) | 3.6 |
| 4c | 3.19 | 3.56 | 3.63 | -a) | 3.75b) | 3.78b) |
| 5a | 4.45b) | 4.1 | 3.64 | -a) | -a) | 3.59 |
| 5b | 3.8 | 4.23b) | 3.67 | -a) | -a) | 3.56 |
| 5c | 3.97 | 4.13 | 3.74 | 3.91b) | -a) | 3.65 |
| 6a | 3.85 | 4.2 | 3.76b) | 3.74 | -a) | 3.67b) |
| 6b | 4.1 | 3.85 | -a) | -a) | 3.55b) | -a) |
| 6c | 4.28b) | 4.65b) | 3.75b) | 4.30b) | -a) | -a) |
a) This target is not included in the top 300 targets for this compound; b) Underlined values represent the highest fit score to each enzyme
Table 3: Fit scores of the tested compounds against the top six targets
Targets proposed by PharmMapper are employed in cancer therapy in a diversity of approaches [8-10,28-30]. As a result, we studied the potential interaction of the synthesized compounds 4-6(a-c) against these six targets which are involved in cancer.
Docking study
The six potential targets proposed by pharmacophore mapping approach were used to investigate their interaction with the designed compounds. The target compounds 4-6(a-c) were comparatively evaluated in terms of estimated free energy of binding (kcal/mol), and inhibition constant Ki (uM) to the proposed enzymes and the results are listed in Table 4. The binary complex of the enzyme coupled with its natural ligand was used as a reference for docking and modeling in this investigation. Docking simulations were carried out with the aid of Docking Server [31]. Compounds showing the best docking score with the target enzymes are 4b and 4c making them possible drug candidates.
| Est. Free Energy of Binding With Different Enzymes (kcal/mol) (Est. Inhibition Constant Ki (uM)) | ||||||
|---|---|---|---|---|---|---|
| Comp. No. | Cathepsin K (1TU6) | Vitamin D3receptor (1DB1) | Dual specificity mitogen-activated protein kinase kinase 1 (1S9J) | Proto-oncogene tyrosine-protein kinase Src (1Y57) | Epidermal growth factor receptor (1XKK) | Leukotriene A4 hydrolase (1GW6) |
| 4a | -6.3 | -6.89 | -6.76 | -6.49 | -7.05 | -6.52 |
| -24.11 | -8.93 | -11.1 | (17.40 | (6.84 | (16.71) | |
| 4b | -6.44 | -7.39 | -7.85 | -8.19 | -7.4 | -6.81 |
| -18.96 | (3.83)a) | (1.76)a) | (0.984)a | (3.79)a) | (10.18)a) | |
| 4c | -6.03 | -7.63 | -7.68 | -7.82 | -7.19 | -6.99 |
| -38.26 | (2.55)a) | -2.33 | (1.86)a) | (5.35)a) | (7.56)a) | |
| 5a | -6.09 | -5.95 | -7.75 | -7.11 | -5.33 | -5.14 |
| -34.17 | (43.82) | (2.10) | (6.13) | (124.9) | (170.4) | |
| 5b | -6.48 | -4.27 | -7.9 | -7.26 | -6.12 | -4.1 |
| (17.67)a) | (744.1) | (1.62)a) | -5.31 | (32.41) | (996.1) | |
| 5c | -6.36 | -5.43 | -7.58 | -6.67 | -6.72 | -5.31 |
| -21.79 | (103.9) | (2.77) | (12.91) | (11.95 | (129.1) | |
| 6a | -6.47 | -2.5 | -6.45 | -7.41 | -5.56 | 3.96 |
| -18.24 | (14660) | (18.84) | (3.73) | (84.59) | (1260) | |
| 6b | -6.42 | -3.57 | -7.65 | -7.59 | 6.08 | -4.01 |
| -19.55 | -2410 | (2.45) | (2.73) | (34.75) | (1150) | |
| 6c | -6.58 | -3.27 | -6.72 | -7.24 | -6.91 | -4.27 |
| (15.10)a) | -3980 | (11.88) | (4.96) | (8.58 | (737.76) | |
| -3.69 | -8.66 | -5.15 | -7.95 | -11.11 | -7.89 | |
| Ref. Lig. | -1990 | -0.452 | (166.8) | (1.49) | (0.007) | (1.64) |
a) Underlined values represent the highest affinity to each enzyme
Table 4: Estimated free energy of binding with different targets and inhibition constants of the tested compounds against the top six targets
Experimental
General
All the reagents and solvents were obtained from commercial suppliers, and used without purification. TLC was monitored on Fluka silica gel TLC aluminum cards (0.2 mm thickness) with fluorescent indicator 254 nm using a mixture of petroleum ether/ethyl acetate in various proportions.
Melting points (°C) were recorded using a Fischer-Johns melting point apparatus and are uncorrected. The IR spectra (KBr) were recorded on Mattson 5000 FT-IR spectrophotometer (ѵ in cm-1) in the Microanalytical Unit, Faculty of Science, Mansoura University. 1H and 13C-NMR for compounds 4a, 5a and 6a were recorded on Bruker 500 MHz FT-NMR spectrometer and 1H-NMR spectra for remaining compounds were carried out at the National Research Centre using a Varian Gemini 500 MHz FT-NMR. Deuteriodimethylsulfoxide (DMSO-d6) is used as a solvent with the chemical shift being expressed in δ (ppm) and downfield from tetramethylsilane (TMS) as internal standard.
Electron impact mass spectra (EI-MS), recorded on a Shimadzu GC/MS QP-2010 Plus mass spectrometer, and elemental analyses (in accord with the calculated values) were carried out in the Microanalytical Unit, Faculty of Science, Cairo University. Anticancer evaluation was performed at National Cancer Institute (NCI), Bethesda, Maryland, USA.
General procedure for synthesis of 2-amino-4-(un)substituted phenylthiazoles (2a–c) [19]
A mixture of phenacyl halide (either phenacyl chloride or phenacyl bromide) 1a-c (10 mmol) and thiourea (0.76 g, 10 mmol) in DMF (10 ml) was stirred at room temperature until completion of the reaction (2-3 min). The progress of the reaction was monitored by thin-layer chromatography. On completion of the reaction, the reaction mixture was poured onto crushed ice, treated with an excess of aqueous Na2CO3 solution. The precipitate was separated by filtration and washed with water. The product was pure enough (single spot on TLC) for all practical purposes.
4-Phenylthiazol-2-amine (2a): Yield: 95%; M. p. 146-148°C (lit. M. p. 146°C) [19]; IR (KBr, ѵ, cm-1): 3436 (N–H), 1598, 1539, 1516 (C=N, C=C) .
4-(4-Chlorophenyl)thiazol-2-amine (2b): Yield: 93%; M. p. 161-163°C (lit. M. p. 161°C) [32].
4-p-Tolylthiazol-2-amine (2c): Yield: 94%; M. p. 126-128°C (lit. M. p. 126°C) [32].
Synthesis of 6-(2-chloroethyl)-7-methyl-3-(un)substituted phenyl 5H-thiazolo[3,2-a]pyrimidin-5-ones (3a–c) [20,21]
α-Acetyl-γ-butyrolactone (1.08 ml, 10 mmol) was added slowly to a solution of 2-amino-4-arylthiazole 2a–c (10 mmol) in phosphorous oxychloride (15 ml). The mixture was refluxed for 18 h, allowed to cool and poured onto crushed ice. The crude product was filtered, dried and crystallized from DMF/EtOH.
6-(2-Chloroethyl)-7-methyl-3-phenyl-5H-thiazolo[3,2-a]pyrimidin5-one (3a): Yield: 52%; M. p. 138-141°C (lit. M. p. 136°C) [20]; IR (KBr, ѵ, cm-1): 3079 (CH aromatic), 2960, 2915 (CH aliphatic), 1651 (C=O), 1598, 1539, 1499 (C=N, C=C).
6-(2-Chloroethyl)-3-(4-chlorophenyl)-7-methyl-5H-thiazolo[3,2-a]pyrimidin-5-one (3b): Yield: 55%; M. p. 156-158°C (lit. M. p. 161-163°C) [20]. 6-(2-Chloroethyl)-7-methyl-3-p-tolyl-5H-thiazolo[3,2-a]pyrimidin-5-one (3c): Yield: 48%; M. p. 188-190°C (lit. M. p. 188°C) [20].
General procedure for the synthesis of compounds 4-6(a-c)
An equimolar amount of 6-(2-chloroethyl)-7-methyl-3-(un)substitutedphenyl-5H-thiazolo[3,2-a]pyrimidin-5-one 3a–c (10 mmol) and the appropriate amine (10 mmol) was heated at 90°C in dry DMF (15 ml) containing triethylamine (2 ml) for 18 h. The reaction mixture was cooled to 20°C and poured onto ice-water. The crude product was filtered, dried, and crystallized from DMF/EtOH to yield the desired compounds.
7-Methyl-6-(2-morpholinoethyl)-3-phenyl-5H-thiazolo[3,2-a]pyrimidin-5-one (4a): Yield: 35%; M. p. 140-142°C; 1H-NMR (δ, ppm): 2.33 (t, 2H, -CH2CH2N-), 2.41 (s, 3H, -CH3), 2.44 (t, 2H, -CH2CH2N-), 2.59 (t, 4H, (CH2)2N-morpholine), 3.55 (br s, 4H, (CH2)2O-morpholine), 7.37 (t, 2H, Ar-H), 7.42 (t, 1H, Ar-H), 7.75 (s, 1H, H-thiazole), 7.94 (d, 2H, Ar-H); 13C-NMR (δ, ppm, DMSO-d6): 21.90 (CH3), 29.53 (CH2CH2N), 53.16 ((CH2)2N-morpholine), 56.51 (CH2CH2N), 66.15 ((CH2)2O-morpholine), 102.44, 109.36, 125.78, 127.02, 128.68, 133.93, 137.13 (aromatic C), 154.82 (C7 of thiazolopyrimidine), 159.77 (C=O), 168.13 (-S-C(N)=N- of thiazolopyrimidine); EI-MS (70 eV) m/z (Rel. Int.): 355 (M+ , 24.35), 278 (2.28), 255 (8.80), 192 (4.40), 176 (15.23), 149 (5.31), 134 (20.51), 114 (7.76); Anal. for C19H21N3O2S (355.45) C, H, N.
3-(4-Chlorophenyl)-7-methyl-6-(2-morpholinoethyl)-5H-thiazolo[3,2-a]pyrimi-din-5-one (4b): Yield: 40%; M. p. 134-136°C; 1H-NMR (δ, ppm): 2.29 (t, 2H, -CH2CH2N-), 2.42 (s, 3H, -CH3), 2.47 (t, 2H, -CH2CH2N-), 2.71 (t, 4H, (CH2)2N-morpholine), 3.53 (t, 4H, (CH2)2O-morpholine), 7.46 (d, 2H, Ar-H), 7.63 (s, 1H, H-thiazole), 7.86 (d, 2H, Ar-H); 13C-NMR (δ, ppm, DMSO-d6): 21.51 (CH3), 25.65 (CH2CH2N), 52.78 ((CH2)2N-morpholine), 62.88 (CH2CH2N), 66.75 ((CH2)2O-morpholine), 111.72, 127.04, 130.93, 131.12, 131.16, 133.20 (aromatic C), 154.62 (C7 of thiazolopyrimidine), 159.25 (C3 of thiazolopyrimidine), 160.51 (C=O), 168.13 (-S-C(N)=N- of thiazolopyrimidine); EI-MS (70 eV) m/z (Rel. Int.): 391 (M++2 , 0.83), 389 (M+ , 2.42), 278 (1.28), 275 (6.32), 223 (3.96), 210 (5.78), 168 (13.07), 114 (9.56); Anal. for C19H20ClN3O2S (389.90) C, H, N.
7-Methyl-6-(2-morpholinoethyl)-3-p-tolyl-5H-thiazolo[3,2-a]pyrimidin-5-one (4c): Yield: 31%; M. p. 130-132°C; 1H-NMR (δ, ppm): 2.30 (m, 5H, -CH2CH2N- and Ar-CH3), 2.42 (s, 3H, -CH3), 2.46 (t, 2H, -CH2CH2N-), 2.69 (t, 4H, (CH2)2N-morpholine), 3.39 (t, 4H, (CH2) 2O-morpholine), 7.18 (d, 2H, Ar-H), 7.23 (s, 1H, H-thiazole), 7.63 (d, 2H, Ar-H); 13C-NMR (δ, ppm, DMSO-d6): 20.87 (CH3), 21.11 (CH3), 29.53 (CH2CH2N), 53.14 ((CH2)2N-morpholine), 56.51 (CH2CH2N), 66.10 ((CH2)2O-morpholine), 100.57, 125.49, 127.64, 128.51, 129.06, 129.26, 137.13 (aromatic C), 154.82 (C7 of thiazolopyrimidine), 159.77 (C=O), 168.10 (-S-C(N)=N- of thiazolopyrimidine); EI-MS (70 eV) m/z (Rel. Int.): 369 (M+ , 14.37), 278 (12.23), 255 (14.17), 203 (17.48), 192 (19.81), 190 (95.73), 176 (12.62), 148 (32.43), 114 (16.31); Anal. for C20H23N3O2S (369.48) C, H, N.
N-(4-(2-(7-Methyl-5-oxo-3-phenyl-5H-thiazolo[3,2-a]pyrimidin-6-yl)ethylamino)-phenylsulfonyl)acetamide (5a): Yield: 38%; M. p. 102- 104°C; 1H-NMR (δ, ppm): 1.93 (s, 3H, -COCH3), 2.34 (t, 2H, -CH2CH2NH-), 2.51 (s, 3H, -CH3), 3.43 (t, 2H, -CH2CH2NH-), 5.88 (s, 1H, - CH2CH2NH-Ar), 5.99 (s, 1H, -ArSO2NH-), 6.59 (d, 2H, NH-Ar-H-SO2NH-), 7.38 (t, 2H, H-Ar-thiazole), 7.42 (t, 1H, H-Ar-thiazole), 7.52 (s, 1H, H-thiazole), 7.78 (d, 2H, NH-Ar-H-SO2NH-), 7.94 (d, 2H, H-Ar-thiazole); 13C-NMR (δ, ppm, DMSO-d6): 21.45 (CH3), 25.16 (COCH3), 29.59 (CH2CH2N), 55.77 (CH2CH2N), 101.31, 109.04, 112.59, 125.67, 127.87, 128.39, 128.89, 131.91, 134.31, 137.15 (aromatic C), 148.80 (NH-aromatic C), 156.58 (C7 of thiazolopyrimidine), 162.27 (CO), 168.05 (-S-C(N)=N- of thiazolopyrimidine), 171.04 (COCH3); EI-MS (70 eV) m/z (Rel. Int.): 482 (M+ , 0.20), 439 (0.09), 424 (0.19), 405 (0.04), 348 (0.05), 284 (1.50), 160 (0.96), 134 (8.45); Anal. for C23H22N4O4S2 (482.58) C, H, N.
N-(4-(2-(3-(4-Chlorophenyl)-7-methyl-5-oxo-5H-thiazolo[3,2-a]pyrimidin-6-yl)ethylamino)phenylsulfonyl)acetamide (5b): Yield: 43%; M. p. 126-128°C; 1H-NMR (δ, ppm): 1.92 (s, 3H, -COCH3), 2.31 (t, 2H, -CH2CH2NH-), 2.45 (s, 3H, -CH3), 3.36 (t, 2H, -CH2CH2NH-), 5.72 (s, 1H, -CH2CH2NH-Ar), 5.90 (s, 1H, -ArSO2NH-), 6.44 (d, 2H, NH-Ar-H-SO2NH-), 7.38 (d, 2H, H-Ar-thiazole), 7.63 (s, 1H, H-thiazole), 7.71 (d, 2H, NH-Ar-H-SO2NH-), 7.86 (d, 2H, H-Ar-thiazole); 13C-NMR (δ, ppm, DMSO-d6): 21.75 (CH3), 26.50 (COCH3), 29.54 (CH2CH2N), 59.19 (CH2CH2N), 102.33, 111.66, 112.56, 112.93, 127.05, 128.38, 131.04, 131.09, 133.14 (aromatic C), 147.47 (C3 of thiazolopyrimidine), 152.42 (NH-aromatic C), 159.27 (C7 of thiazolopyrimidine), 160.27 (C=O), 168.78 (-S-C(N)=N- of thiazolopyrimidine), 170.41 (COCH3); EI-MS (70 eV) m/z (Rel. Int.): 518 (M++2 , 1.07), 516 (M+ , 1.52), 473 (1.07), 458 (0.96), 405 (1.67), 348 (1.37), 318 (1.30), 210 (79.80), 194 (4.46), 168 (37.89); Anal. for C23H21ClN4O4S2 (517.02) C, H, N.
N-(4-(2-(7-Methyl-5-oxo-3-p-tolyl-5H-thiazolo[3,2-a]pyrimidin-6-yl)ethylamino)-phenylsulfonyl)acetamide (5c): Yield: 35%; M. p. 152- 154°C; 1H-NMR (δ, ppm): 1.92 (s, 3H, -COCH3), 2.30 (m, 5H, -CH2CH2N- and Ar-CH3), 2.47 (s, 3H, -CH3), 3.32 (t, 2H, -CH2CH2NH-), 5.78 (s, 1H, -CH2CH2NH-Ar, D2O exchangeable), 5.86 (s, 1H, -ArSO2NH-, D2O exchangeable), 6.55 (d, 2H, NH-Ar-H-SO2NH-), 7.13 (d, 2H, H-Ar-thiazole), 7.20 (s, 1H, H-thiazole), 7.49 (d, 2H, NH-Ar-H-SO2NH-), 7.64 (d, 2H, H-Ar-thiazole); 13C-NMR (δ, ppm, DMSO-d6): 20.86 (CH3), 26.50 (COCH3), 30.78 (CH2CH2N), 35.81 (CH3), 50.57 (CH2CH2N), 100.56, 110.59, 112.41, 112.54, 125.48, 127.62, 128.35, 129.04, 129.23 (aromatic C), 149.00 (C3 of thiazolopyrimidine), 149.90 (NH-aromatic C), 158.79 (C7 of thiazolopyrimidine), 162.33 (C=O), 168.09 (-SC-( N)=N- of thiazolopyrimidine), 170.41 (COCH3); EI-MS (70 eV) m/z (Rel. Int.): 496 (M+ , 15.32), 453 (48.65), 405 (63.06), 348 (63.06), 190 (42.34), 174 (72.97), 148 (25.23); Anal. for C24H24N4O4S2 (496.60) C, H, N.
N-Carbamimidoyl-4-(2-(7-methyl-5-oxo-3-phenyl-5H-thiazolo[3,2-a]pyrimidin-6-yl)ethylamino)benzenesulfonamide (6a): Yield: 41%; M. p. 134-136°C; 1H-NMR (δ, ppm): 2.28 (t, 2H, -CH2CH2NH-), 2.51 (s, 3H, -CH3), 3.46 (t, 2H, -CH2CH2NH-), 4.12 (s, 1H, -ArSO2NH-C(NH)-), 4.33 (s, 2H, H2N-C(NH)-), 6.05 (s, 1H, -CH2CH2NH-Ar), 6.58 (d, 2H, NH-Ar-H-SO2NH-), 7.04 (s, 1H, -ArSO2NH-), 7.37 (t, 2H, H-Ar-thiazole), 7.40 (t, 1H, H-Ar-thiazole), 7.63 (s, 1H, H-thiazole), 7.91 (d, 2H, NH-Ar-H-SO2NH-), 7.94 (d, 2H, H-Ar-thiazole); 13C-NMR (δ, ppm, DMSO-d6): 21.49 (CH3), 29.47 (CH2CH2N), 59.79 (CH2CH2N), 101.46, 109.98, 118.31, 125.63, 127.15, 128.61, 129.12, 130.63, 132.01, 137.15 (aromatic C), 148.80 (NH-aromatic C), 157.75 (C7 of thiazolopyrimidine), 159.23 (C(NH)NH2), 161.30 (C=O), 168.17 (-S-C(N)=N- of thiazolopyrimidine); EI-MS (70 eV) m/z (Rel. Int.): 482 (M+ , 0.01), 284 (0.42), 241 (4.24), 238 (0.83), 189 (11.31), 176 (100); Anal. for C22H22N6O3S2 (482.58) C, H, N.
N-Carbamimidoyl-4-(2-(3-(4-chlorophenyl)-7-methyl-5-oxo-5H-thiazolo[3,2-a]pyrimidin-6-yl)ethylamino)benzenesulfonamide (6b): Yield: 49%; M. p. 148-150°C; 1H-NMR (δ, ppm): 2.31 (t, 2H, -CH2CH2NH-), 2.46 (s, 3H, -CH3), 3.05 (t, 2H, -CH2CH2NH-), 4.12 (s, 1H, - ArSO2NH-C(NH)-), 4.53 (s, 2H, H2N-C(NH)-), 5.99 (s, 1H, -CH2CH2NH-Ar), 6.54 (d, 2H, NH-Ar-H-SO2NH-), 7.06 (s, 1H, -ArSO2NH-), 7.38 (d, 2H, H-Ar-thiazole), 7.63 (s, 1H, H-thiazole), 7.78 (d, 2H, NH-Ar-H-SO2NH-), 7.89 (d, 2H, H-Ar-thiazole); 13C-NMR (δ, ppm, DMSO-d6): 20.98 (CH3), 29.53 (CH2CH2N), 59.20 (CH2CH2N), 102.34, 108.65, 109.46, 111.68, 127.03, 127.24, 128.50, 128.80, 131.06 (aromatic C), 147.48 (C3 of thiazolopyrimidine), 148.59 (NH-aromatic C), 159.03 (C7 of thiazolopyrimidine), 159.41 (C(NH)NH2), 160.21 (C=O), 168.36 (- S-C(N)=N- of thiazolopyrimidine); EI-MS (70 eV) m/z (Rel. Int.): 518 (M++2 , 0.31), 516 (M+ , 0.27), 481 (0.26), 318 (0.34), 275 (0.59), 272 (0.37), 223 (1.57), 210 (100); Anal. for C22H21ClN6O3S2 (517.02) C, H, N.
N-Carbamimidoyl-4-(2-(7-methyl-5-oxo-3-p-tolyl-5H-thiazolo[3,2-a]pyrimidin-6-yl)ethylamino)benzenesulfonamide (6c): Yield: 35%; M. p. 168-170°C; 1H-NMR (δ, ppm): 2.29 (m, 5H, -CH2CH2N- and Ar-CH3), 2.40 (s, 3H, -CH3), 3.32 (t, 2H, -CH2CH2NH-), 4.10 (s, 1H, - ArSO2NH-C(NH)-, D2O exchangeable), 4.53 (s, 2H, H2N-C(NH)-, D2O exchangeable), 5.99 (s, 1H, -CH2CH2NH-Ar, D2O exchangeable), 6.55 (d, 2H, NH-Ar-H-SO2NH-), 6.98 (s, 1H, -ArSO2NH-), 7.13 (d, 2H, H-Ar-thiazole), 7.20 (s, 1H, H-thiazole), 7.64 (d, 2H, H-Ar-thiazole), 7.79 (d, 2H, NH-Ar-H-SO2NH-); 13C-NMR (δ, ppm, DMSO-d6): 20.81 (CH3), 21.10 (CH3), 30.78 (CH2CH2N), 59.02 (CH2CH2N), 100.55, 112.30, 125.48, 125.57, 127.27, 128.35, 129.03, 129.22, 129.32, 137.15 (aromatic C), 150.00 (NH-aromatic C), 158.00 (C7 of thiazolopyrimidine), 158.60 (C(NH)NH2), 162.33 (C=O), 168.08 (-S-C(N)=N- of thiazolopyrimidine); EI-MS (70 eV) m/z (Rel. Int.): 496 (M+ , 40.00), 481 (34.84), 255 (7.10), 252 (37.42), 203 (2.58), 190 (50.97); Anal. for C23H24N6O3S2 (496.60) C, H, N.
Full NCI 60 cell panel in vitro anticancer assay
The synthesized compounds 4-6(a-c) were subjected to the National Cancer Institute (NCI) in vitro disease-oriented human cells screening panel assay for in vitro antitumor activity according standard procedure which is previously reported [22-25].
Conclusion
On the basis of results obtained, it was found that the synthesized compounds showed observed activity against Renal UO-31 cancer cell line with cell growth inhibition 36 to 48% at a dose of 10 M. Compounds 5c, 4a, and 4b proved to be the most active members in this study. They showed moderate potency over certain cancer cell lines. From the obtained results, it is clear that substituents affect the activity of compounds in different series. The in silico studies together with in vitro anticancer activity make 5c, 4a, and 4b promising lead compounds for development of more potent anticancer agents.
Acknowledgement
We are thankful to the National Cancer Institute (NCI), Bethesda, Maryland, USA, for performing the anticancer evaluation over the 60-cancer cell line panel.
References
- A.R. Peter, A.P. Singh, C.K.V. Lucile, F.A. Marie, F.R. Jeremy, Fused thiazole derivatives as kinase inhibitors, EP 2035436 B1.
- B.S. Holla, B.S. Rao, B.K. Sarojini, P.M. Akberali, Eur. J. Med. Chem., 2004, 39, 777-783.
- A.M. Mohamed, A.E. Amr, M.A. Alsharari, H.R.M. Al-Qalawi, M.O. Germoush, M.A. Al-Omar, Am. J. Biochem. & Biotech., 2011, 7, 43-54.
- A.A. Abu-Hashem, M.M. Youssef, H.A.R. Hussein, J. Chin. Chem. Soc., 2011, 58, 41-48.
- T.P. Selvam, V. Karthick, P.V. Kumar, M.A. Ali, Drug Discov. Ther., 2012, 6, 198-204.
- A.R. Ali, E.R. El-Bendary, M.A. Ghaly, I.A. Shehata, Eur. J. Med. Chem., 2013, 69, 908-919.
- A.R. Ali, E.R. El-Bendary, M.A. Ghaly, I.A. Shehata, Eur. J. Med. Chem., 2014, 75, 492-500.
- C. Gridelli, F.D. Marinisb, M.D. Maioc, D. Cortinovisd, F. Cappuzzoe, T. Mokf, Lung Cancer.,2011, 71, 249-257.
- J.F. Ohren, H. Chen, A. Pavlovsky, C. Whitehead, E. Zhang, P. Kuffa, C. Yan, P. McConnell, C. Spessard, C. Banotai, W.T. Mueller, A. Delaney, C. Omer, J.S.S. Leopold, D.T. Dudley, I.K. Leung, C. Flamme, J. Warmus, M. Kaufman, S. Barrett, H. Tecle, C.A. Hasemann, Nature Struc. Mol. Biol., 2004, 11, 1192-1197.
- R. Thaimattam, P.R. Daga, R. Banerjeea, J. Iqbal, Bioorg. Med. Chem., 2005, 13, 4704-4712.
- G.L. Patrick, Anticancer Agents'' In: An introduction to medicinal chemistry, G.L. Patrick (Edi.), 4th (Edn.), Oxford university press, 2009, 519-578.
- M.S. Al-Said, M.S. Bashandy, S.I. Al-qasoumi, M.M. Ghorab, Eur. J. Med. Chem., 2011, 46, 137-141.
- S.I. Alqasoumi, A.M. Al-Taweel, A.M. Alafeefy, E. Noaman, M.M. Ghorab, Eur. J. Med. Chem., 2010, 45, 738-744.
- T. Owa, T. Okauchiy, K. Yoshimatsu, N.H. Sugi, Y. Ozawa, T. Nagasu, N. Koyanagi, T. Okabe, K. Kitoh, H. Yoshino, Bioorg. Med. Chem. Lett., 2000, 10, 1223-1226.
- P.B. Chapman, A. Hauschild, C. Robert, J.B. Haanen, P. Ascierto, J. Larkin, R. Dummer, C. Garbe, A. Testori, M. Maio, D. Hogg, P. Lorigan, C. Lebbe, T. Jouary, D. Schadendorf, A. Ribas, S.J. O’Day, J.A. Sosman, J.M. Kirkwood, A.M.M. Eggermont, B. Dreno, K. Nolop, J. Li, B. Nelson, J. Hou, R.J. Lee, K.T. Flaherty, G.A. McArthur, N. Engl. J. Med., 2011, 364, 2507-2516.
- K.A. azi, H. Lawrence, W.C. Guida, M.L. McLaughlin, G.M. Springett, N. Berndt, R.M.L. Yip, S.M. Sebti, Cell Cycle., 2009, 8, 1940-1951.
- M.M. Ghorab, F.A. Ragab, H.I. Heiba, A.A. Bayomi, Life Sci. J., 2013, 10(4), 2184-2192.
- Z. Wang, Y. Qin, P. Wang, Y. Yang, Q. Wen, X. Zhang, H. Qiu, Y. Duan, Y. Wang, Y. Sang, H. Zhu, Eur. J. Med. Chem., 2013, 66, 1-11.
- S.N. Dighe, P.K. Chaskar, K.S. Jain, M.S. Phoujdar, K.V. Srinivasan, ISRN Org. Chem.,2011, 2011, 1-6.
- R. Foguet, L. Anglada, A. Sacristan, J. Castello, J.A. Ortiz, Chem. Abstr., 1997, 126, 89392v.
- F.M. Awadallah, Sci Pharm.,2008, 76, 415-438.
- www.dtp.nci.nih.gov
- M.R. Grever, S.A. Schepartz, B.A. Chabner, Semin. Oncol., 1992, 19, 622-638.
- A. Monks, D. Scudiero, P. Skehan, R. Shoemaker, K. Paull, D. Vistica, C. Hose, J. Langley, P. Cronise, A.V. Wolff, J. Natl. Cancer Inst., 1991, 83, 757-766.
- M.R. Boyd, K.D. Paull, Drug Dev. Res., 1995, 34, 91-109.
- http://www.organic-chemistry.org/prog/peo.
- X. Liu, S. Ouyang, B. Yu, K. Huang, Y. Liu, J. Gong, S. Zheng, Z. Li, H. Li, H. Jiang, Nucleic Acids Res., 2010, 38, 609-614.
- D.G. Barrett, J.G. Catalano, D.N. Deaton, A.M. Hassell, S.T. Long, A.B. Miller, L.R. Miller, L.M. Shewchuk, K.J. Wells-Knecht, D.H. Willard, L.L. Jr. Wright, Bioorg. Med. Chem. Lett., 2004, 14, 4897-4902.
- C. Carlberg, F. Molnár, Curr. Top. Med. Chem., 2012, 12, 528-547.
- V. Sandanayaka, B. Mamat, N. Bhagat, L. Bedell, G. Halldorsdottir, H. Sigthorsdottir, P. Andrésson, A. Kiselyov, M. Gurney, J. Singh, Bioorg. Med. Chem. Lett.,2010, 20, 2851-2854.
- Z. Bikadi, S. Kovacs, L. Demko, E. Hazai, DockingServer (www.dockingserver.com), Virtua Drug Ltd., Budapest, Hungary, 2007.
- L.C. King, R.J. Hlavacek, J. Am. Chem. Soc., 1950, 72, 3722-3725.



