Research Article - Der Pharma Chemica ( 2019) Volume 11, Issue 5
Theoretical Bio-Estimation of 1,2,3-triazolo[4,5-d]pyrimidine Hybrids using DFT, Multiple Linear Regression (MLR) and Docking Approaches
Oyebamiji AK1,2 and Semire B2*2Computational Chemistry Laboratory, Department of Pure and Applied Chemistry, Ladoke Akintola University of Technology, P.M.B. 4000, Ogbomoso, Oyo State, Nigeria
Semire B, Computational Chemistry Laboratory, Department of Pure and Applied Chemistry, Ladoke Akintola, University of Technology, P.M.B. 4000, Ogbomoso, Oyo State, Nigeria, Email: bsemire@lautech.edu.ng
Published: 01-Jan-0001
Abstract
26 sets of molecular compounds were studied for anti-esophageal cancer activity. B3LYP/6-31+G* basis set was used for Quantum chemical calculation and the obtained molecular descriptors were used for QSAR studies via Gretl software. Thus, the developed model predicted efficiently and it was used to predict the cytotoxicity of the proposed compounds. Furthermore, molecular docking study was carried out on esophageal cancer cell line (2leo) and it was observed that A13 inhibited more than all the studied compounds. Also, all the studied compounds were more effective than the standard (5-FU).
Keywords
1,2,3-triazolo[4, 5-d]pyrimidine hybrids, DFT, QSAR, Molecular docking.
Introduction
Recently, the broad uses of antibiotics against microorganism have brought about a new search for more effective drug-like molecules and modification of the existing antibiotics. Cancer, a frenzied cell growth, is positioned high amidst every disease attacking humans and have been reported to be the cause of death of several human beings [1]. Despite the continuous effort of scientist over the world to overcome cancer, the more the number of people with cancer upturns [2]. Globally, its position as an agent of death still remains the second. According to Fridlender et al., and Gali-Muhtasib et alcancer could be caused by malfunction of basic cellular processes in human body [3,4].
Also, esophageal cancer is a deadly tumor. It is ranked to be one of the top five most dreaded tumor ever recorded [5]. It is majorly sub-divided into two, esophageal squamous-cell carcinoma (ESCC) and esophageal adenocarcinoma (EA). ESCC is a common malevolence that is rated to be fourth leading cause of deaths in Asia region [6]. It diagnosis is poor and the cure for this malignant disease is still a serious threat to medicinal world [7,8].
In the designing and development of drug-like molecules, the role play by heterocyclic compounds is very vital. 1, 2, 3-triazolo[4,5-d]pyrimidine analogues as heterocyclic molecules have two major active compounds i.e. triazole and pyrimidine. Both triazole and pyrimidine are important class of molecular compounds with reputable bio-activities and this has drawn the attention of researchers over the world. Triazole and Pyrimidine derivatives possess many biological activities such as antimicrobial [9,10], anticancer [11], antioxidant [12], anti-inflammatory [13,14], diuretics [15] and analgesics [16,17].
As shown in Figure 1, twenty-six selected molecular compounds used in this work were optimized using density functional theory. The compounds used were randomly selected from two experimental research work and were subjected to further studies. Compounds A1-A15 as shown in Figure 1 were taken from Li et al., [18] and compound A16-A26 were chosen from Li et al., [19]. The optimized molecular compounds are 3-benzyl-7-(2-(2-chlorobenzylidene)hydrazinyl)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidine (A1), 3-benzyl-7-(2-(4-nitrobenzylidene)hydrazinyl)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidine (A2), 4-((2-(3-benzyl-5-(propylthio)-3H-[1,2,3]triazole-[4,5-d]pyrimidin-7-yl)hydrazono)methyl)-N,N-dimethylaniline (A3), 7-(2-((1H-indol-3-yl)methylene)hydrazinyl)-3-benzyl-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidine (A4), 1-((2-(3-benzyl-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-7-yl)hydrazono)methyl)naphthalen-2-ol (A5), 2-(1-(2-(3-benzyl-5-(propylthio)-3H-[1,2,3]-triazolo[4,5-d]-pyrimidin-7-yl)-hydrazono)-ethyl)phenol (A6), 3-benzyl-7-(2-(1-(4-bromophenyl)ethylidene)hydrazinyl)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidine (A7), 2-(1-(2-(3-(3-chlorobenzyl)-5-(propylthio)-3H-[1,2,3]-triazolo[4,5-d]pyrimidin-7-yl)-hydrazono)-ethyl)phenol (A8), 2-(1-(2-(3-(4-chlorobenzyl)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-7-yl)hydrazono)ethyl)-phenol (A9), 2-(1-(2-(3-(furan-2-ylmethyl)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]-pyrimidin-7-yl)-hydrazono)ethyl)phenol (A10), 2-(1-(2-(3-(4-hydroxybenzyl)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-7-yl)hydrazono)ethyl)-phenol (A11), 3-benzyl-5-(prop-2-yn-1-ylthio)-7-(2-(3,4,5-trimethoxy-benzylidene)hydrazinyl)-3H-[1,2,3]triazolo[4,5-d]pyrimidine (A12), 1-((2-(3-benzyl-5-(prop-2-yn-1-ylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-7-yl)hydrazono)methyl)naphthalen-2-ol (A13), 2-(1-(2-(3-benzyl-5-(prop-2-yn-1-ylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-7-yl)hydrazono)-ethyl)phenol (A14), 3-benzyl-5-(benzylthio)-7-(2-(3,4,5-trimethoxybenzylidene)hydrazinyl)-3H-[1,2,3]triazolo[4,5-d]pyrimidine (A15), 1-(2-((3-Phenethyl-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-7-yl)ami-no) ethyl)-3-(3-(trifluoromethyl)phenyl)thiourea (A16), 1-(3-Chlorophenyl)-3-(2-((3-phenethyl-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d] pyrimidin-7-yl)amino)ethyl)thiourea (A17), 1-(4-Chlorophenyl)-3-(2-((3-phenethyl-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d] pyrimidin-7-yl)amino)ethyl)thiourea (A18), 1-(4-Butylphenyl)-3-(2-((3-phenethyl-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d] pyrimidin-7-yl)amino)ethyl)thiourea (A19), 1-(2-((3-Phenethyl-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-7-yl)amino) ethyl)-3-(pyridin-2-yl)thiourea (A20), 1-(2-((3-Phenethyl-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-7-yl)amino) ethyl)-3-(pyridin-3-yl)thiourea (A21), 1-Benzyl-3-(2-((3-phenethyl-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-7-yl)amino)ethyl)thiourea (A22), 1-(2-((3-Cyclopentyl-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-7-yl)amino)ethyl)-3-(pyridin-3-yl)thiourea (A23), 1-(2-((3-(2-hydroxyethyl)-5-(propylthio)-3H-[1,2,3]-triazolo[4,5-d]-pyrimidin-7-yl) amino)ethyl)-3-(pyridin-3-yl)thiourea (A24), 1-(2-((3-benzyl-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-7-yl)amino) ethyl)-3-(pyridin-3-yl)thiourea (A25), 1-(2-((3-(2-(1H-indol-3-yl)ethyl)-5-(propylthio)-3H-[1,2,3]triazolo[4,5-d] pyrimidin-7-yl)amino)ethyl)-3-(pyridin-3-yl)thiourea (A26).
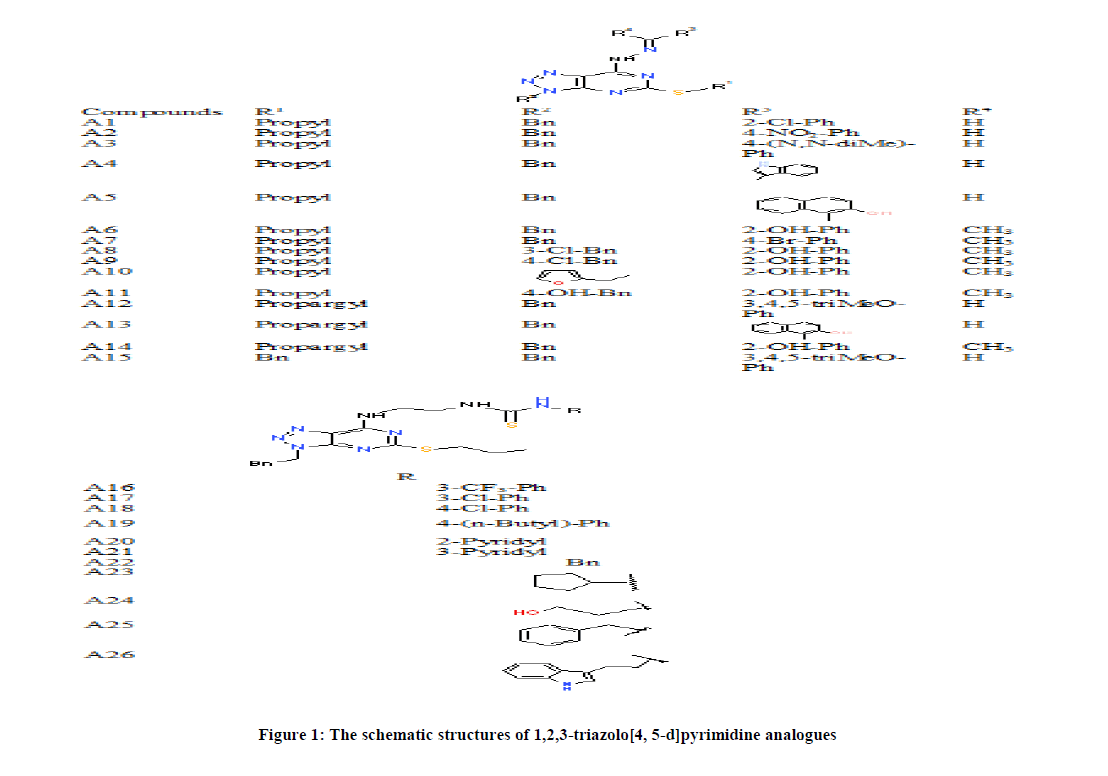
Figure 1: The schematic structures of 1,2,3-triazolo[4, 5-d]pyrimidine analogues
Thus, this work is aimed at developing effective QSAR model via multiple linear regression (MLR) method using the calculated molecular descriptors of 1,2,3-triazolo[4,5-d]pyrimidine Hybrids as well as examining the non-bonding interactions between 1,2,3-triazolo[4,5-d]pyrimidine Hybrids and Esophageal cancer cell line (PDB ID: 2leo) [20] via molecular docking.
Materials and Methods
Methodology
Quantum chemical method via Density functional theory method (6-31+G* basis set) was used for optimization of the studied compounds (1, 2, 3-triazolo[4, 5-d]pyrimidine derivatives) in gas phase and the energy calculation was executed in water using Spartan 14 [21]. As shown in Table 1, several parameters were obtained from quantum chemical study and were used further study in this work. Moreover, the obtained calculated descriptors were used to develop QSAR model in order to predict inhibition concentration (IC50). The calculated molecular descriptors were highest occupied molecular orbital energy (EHOMO), the lowest unoccupied molecular orbital energy (ELUMO), band gap (eV), Ovality, dipole moment (Debye), log P, and molecular weight (amp). The developed QSAR model for multiple linear regression was achieved by using Gretl [22].
| Observed IC50 | Fitted IC50 | |
|---|---|---|
| A1* | -0.79 | -0.94 |
| A2 | -0.82 | -0.88 |
| A3 | -1.30 | -1.32 |
| A4 | -0.93 | -0.94 |
| A5 | -0.96 | -0.89 |
| A6* | -1.04 | -1.00 |
| A7 | -0.66 | -0.79 |
| A8 | -1.11 | -0.99 |
| A9 | -1.05 | -1.13 |
| A10 | -0.76 | -0.66 |
| A11 | -1.20 | -1.26 |
| A12* | -0.81 | -0.89 |
| A13 | -0.81 | -0.84 |
| A14 | -0.72 | -0.66 |
| A15 | -0.64 | -0.66 |
| A16 | -1.24 | -1.24 |
| A17 | -1.07 | -0.98 |
| A18* | -1.02 | -1.02 |
| A19 | -1.27 | -1.24 |
| A20 | -0.73 | -0.80 |
| A21 | -0.69 | -0.78 |
| A22 | -1.13 | -1.08 |
| A23* | -1.09 | -1.25 |
| A24 | -1.26 | -1.22 |
| A25 | -1.03 | -1.01 |
| A26* | -0.90 | -1.46 |
| *denote test set | ||
Table 1: Observed IC50 and predicted IC50
Moreover, several statistical parameters such as squared correlation coefficient (R2), adjusted squared correlation coefficient R2 adj, cross validation (CV.R2) (equations 1 and 2) and the significance level (p-value) [23] were used for QSAR model validation. Also, non-bonding interactions between the studied ligand-receptor complex as well as the binding energy were observed using docking approach. In this study, the grid dimension (grid size) used for the studied receptor (2leo) is 28 × 24 × 28 Å and the grid centre used for X, Y and Z coordinate are -3.266 Å, 0.702 Å and 0.427 Å respectively. Inhibition constant (Ki) was calculated using equation 3.

The R2 adjusted could be calculated using equation (2)


Conclusion
Bioactivity of 26 sets of 1, 2, 3-triazolo[4, 5-d]pyrimidine derivatives were studied by observing the calculated electronic descriptors using density functional theory method via 6-31+G(d,p) basis set. The selected calculated molecular descriptors which described anti-Esophageal cancer activity of 1, 2, 3-triazolo[4, 5-d]pyrimidine analogues were used in QSAR study. The QSAR method used was predictive and the docking study was also observed to show the non-bonding interaction as well as the binding affinity between the studied compounds and the receptor. Thus, in the docking study, 1-((2-(3-benzyl-5-(prop-2-yn-1-ylthio)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-7-yl)hydrazono)methyl)naphthalen-2-ol (A13) inhibited more effectively than other compounds.
References
- C.S. Lee, J. Baek, S.Y. Han, Molecules., 2017, 22(29), 1411,
- S.A. Shin, S.Y. Moon, W.Y. Kim, S.M. Paek, H.H. Park, C.S. Lee, Int. J. Mol. Sci., 2018, 19, 2651.
- M. Fridlender, Y. Kapulnik, H. Koltai, Front. Plant Sci., 2015, 6, 799.
- H. Gali-Muhtasib, R. Hmadi, M. Kareh, R. Tohme, N. Darwiche, Apoptosis.,2015, 20, 1531-1562.
- Y. Yang, Z. Tian, Y. Ding, X. Li, Z. Zhang, L. Yang, F. Zhao, F. Ren, and R. Guo, J. Immunol. Res., 2018, 2018, 1-10.
- R.L., Siegel, K.D. Miller, A. Jemal, CA Cancer J. Clin., 2016, 66, 7-30.
- W. Chen, Chin. J. Cancer Res.,2015, 27, 1.
- S. Ohashi, Gastroenterol., 2015, 149, 1700-1715.
- N.K. Basu, F.L. Rose, J. Chem. Soc., 1963, 5660.
- M.A. El-Dawy, A.B. Hozzoa, J. Pharm Sci., 1983, 72, 45.
- F. Clemence, C. Moushart, P. Mackeiwicz, F. Delkevaller, Eur. Pat. Appl. Ep., 1986, 176, 444.
- L.G. Cherkovskaya, E.G. Knysh, G.K. Rogul’chenko, M.S. Drogovoz, S.I. Sal’nikova, I. steblyuk, Farm. Zh (kiev), 1989, 5, 67.
- F. Lazrak, E.M. Essassi, Y. K. Rodi, K. Misbahi, M. Pierrot, Phosphorus, Sulfur, and Silicon, 2004, 179, 1799-1808.
- F. Lazrak, N.H. Ahabchane, A. Keita, E.M. Essassi, M. Pierrot, Indian J. Chem.- Section B Org. Med. Chem., 2002, 41(4), 821-825.
- H. Kumar, A.S. Javed, A.S. Khan, M. Amir, Eur. J. Med. Chem., 2008, 43, 2688-2698.
- R. Gupta, A.K. Gupta, S. Paul, Indian J. Chem., Sect. B, 2000, 39, 847.
- M.T.M. El Wassimy, M. Abdel-Rahman, A.B.A.G. Ghattas, O.A.A. Abdallah, Phosphorus. Sulfur & Silicon, 1992, 70, 99-108.
- Z.H. Li, D.X. Yang, P.F. Geng, J. Zhang, H.M. Wei, B. Hu, Q Guo, X.H. Zhang, W.G. Guo, B. Zhao, B. Yu, L.Y. Ma, H.M. Liu, Eur. J. Med. Chem., 2016, 124, 967e980.
- Z.H. Li, X.Q. Liu, T.Q. Zhao, P.F. Geng, W.G. Guo, B. Yu, H.M. Liu, Eur. J. Med. Chem.,2017,139, 741e749.
- Y. Feng, Y. Geng, T. Zhou, J. Wang, J. Biomol. Nmr., 2012, 53, 65-70.
- Spartan 14, Wave function, INC, Irvine, CA, 2015.
- http://gretl.sourceforge.net/
- M. Ghamali, S. Chtita, A. Ousaa, B. Elidrissi, M. Bouachrine, T. Lakhlifi, Journal of Taibah University for Science,2017, 11, 1-10.



