Research Article - Der Pharma Chemica ( 2018) Volume 10, Issue 11
An Efficient and Alternative Method for Synthesis of Tofacitinib
Srishylam V1*, Devanna1 and Naveen Mulakayala2
1Department of Chemistry, Jawaharlal Nehru Technological University Anantapur, Ananthapuramu, Andhra Pradesh, India
2Clearsynth Labs Ltd. IDA mallapur, Nacharam, Hyderabad-500076, India
- *Corresponding Author:
- Srishylam V
Department of Chemistry
Jawaharlal Nehru Technological University Anantapur
Ananthapuramu, Andhra Pradesh, India
Abstract
An effective synthetic process for the preparation of 3-Benzyl-6-methyl-7-oxa-3-aza-bicyclo[4.1.0]heptanes 7 and 3-Benzyl-6-methyl-3,7-diaza-bicyclo[ 4.1.0]heptane-7-carboxylic acid tert-butyl ester 9 has been developed from simple synthetic transformations, starting with extraordinarily simple materials (4-picoline and benzyl chloride) in five steps through reduction and epoxidation followed by ring opening, providing a new and convenient access to the key intermediates of Tofacitibin (CP-690,550).The process developed is highly efficient and can be applied for large scale synthesis also.
Keywords
Piperidines, Oxirane, Aziridine, Tofacitinib (CP-690,550).
Introduction
The piperidine moiety is one of the privileged structures1 in medicinal chemistry [1-6]. In particular, 3-aminopiperidines appear quite frequently in pharmaceutically compounds [7-18]. An additional substituent in the 4-position of such compounds can be introduced by nucleophilic ring opening of 3,4-aziridinopiperidines [19,20]. The compound Tofacitinib, (3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl) amino)piperidin-1-yl)-3-oxopropanenitrile) is a small molecule designed by Pfizer, also known as CP-690,550 with the chemical structure of substituted piperidine linked to a deazapurine core, as shown in Figure 1, And it is a promising immunosuppressant compound, less toxic than predecessors and very effective, orally active, and described as a JAK3 specific inhibitor, which has demonstrated efficacy against psoriasis, rheumatoid arthritis, Chron’s disease, kidney transplant rejection, and ulcerative colitis [21-24]. Due to its highly potent biological activities, many groups have tried to develop an efficient and alternative strategies for the synthesis of Tofacitinib, In this context, we considered orthogonally protected tert-butyl 4-benzyl-1-methyl-4,7-diaza-bicyclo[4.1.0]heptane-7-carboxylate 9 and 4-benzyl-1-methyl-7-oxa-4-aza-bicyclo[ 4.1.0]heptaneto7 [25] are an extraordinarily useful building blocks for the preparation of 4-substituted 3-aminopiperidine derivatives. Herein, we wish to report a straightforward synthesis of these racemic compounds 7 and 10, are the key intermediates for making tofacitinib starting from readily available starting materials.
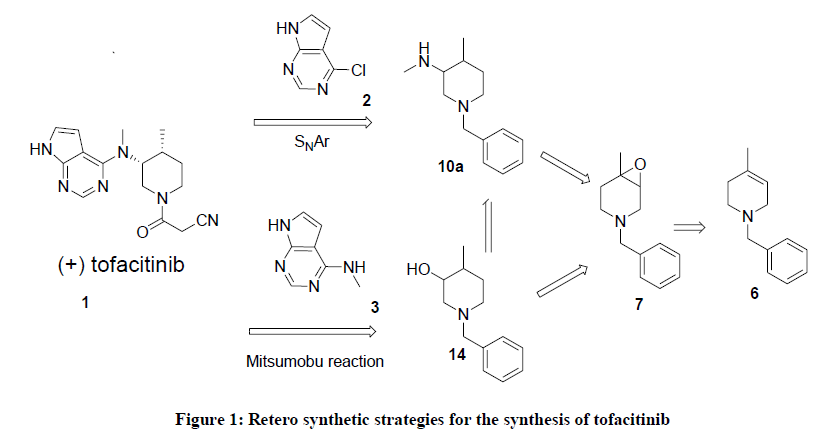
Figure 1: Retero synthetic strategies for the synthesis of tofacitinib
Although several methods for the synthesis of tofacitinib are known in the literature, asymmetric synthesis is rare [26-28]. Most of reported syntheses require resolution of racemiic 3,4-disubstituted piperidines 11 [29,30] or 14 [31], chiral 3,4-disubstituted piperidine 11 is reacted with 6-chloro-7-deazapurine 2 through an SNAr mechanism or a Mitsunobu reaction [32,33] of 14 with 6- amino-7-deazapurine 3 to give the advanced intermediates for the synthesis of (+)-tofacitinib (Figure 1).
Results and Discussion
Based on its pharmaceutical importance, the synthesis of tofacitinib was extensively studied, and we propose a complete new approach for the synthesis of this new immunosuppressive compound. From a retrosynthetic point of view, orthogonally protected aziridine 9 and 1-Benzyl-4- methyl-piperidin-3-ol [31] 14 can be synthesized from olefin 6 via the corresponding epoxide 7 (Figure 1) [25].Tetrahydropyridine derivative 6 might be prepared by addition of methyllithium to 1-Benzyl-piperidin-4-one, followed by tertiary chloride formation and elimination provided olefin 6 with 60% yield [31]. Alternatively, an aqueous Diels-alder reaction produced the material from isoprene and favored route to intermediate 6 [34], however, these routes have some drawbacks like long reaction time, low yields, handling of methyl lithium and isolation of product from diels-alder reaction.
As an alternative to above reactions, we envisioned the reduction of 4-picoline to give 4-methyl- tetrahydropyridine, probably the most inexpensive approach to this target is the solvent free benzylation of 4-picoline itself and subsequent treatment of the benzylpyridinium chloride with sodium borohydride in protic solvents. This reaction was previously reported in the literature to give 73% yield [35]. We were able to isolate compound 6 in 90% yield. Attempts at subsequent epoxidation of tertiary amine 6 were unfortunately not chemoselective. The conversion was practically quantitative, but the purification of epoxide 7 became very difficult because of its instability, gave the respective amine-N-oxide, 1-benzyl-4-methyl 3,4-dihydroxypiperidine as major products after chromatography over both silica gel and aluminia. By careful adjustment of reaction condition and detailed chromatographic monitoring, In the overall yield of 15% were obtained the target epoxide [25] using trifluoroacetic acid, trifluoroacetic anhydride and H2O2 in dichloromethane.All the above results indicated that the necessity for the synthetic conditions and appropriate epoxidizing agent to successful preparation of 1-benzyl-4-methyl-3,4-epoxy piperidine. We turned our attention to the exploration of epoxidation (Table 1) protocol by the addition of different oxidents, acids and solvents. Among all the protocol condition tertiary amine 6 could be converted into trifluoroacitic acid salt with excess TFA at 25°C-30°C in dry dichloromethane, Subsequent epoxidation with m-chloro-peroxybenzoic acid addition at 0°C-5°C portion wise gave oxirane 7 with high selectivity and in almost quantitative yield (85%-90%) after 2-3 h at 25°C-30°C.The crude material was pure enough to be submitted to the next step without purification. The ring opening with sodium azide was achieved in acetic acid water, and yielded a 93: 07 mixture of two regioisomers 8a and 8b, respectively, in an overall yield of 85%. These isomers could, but did not need to be separated by column chromatography. Staudinger reaction of this mixture of azides 8a and 8b was performed with triphenylphosphane in absolute toluene at 100°C for 10 h. As reported in the literature for the respective cyclohexane derivative [36], activation of the hydroxy group as recommended by others [37], was not necessary, since it directly reacts with the iminophosphorane to form the aziridine and triphenylphosphane oxide (Ph3P=O). It turned out that the parent aziridine (without Boc protection) was difficult to isolate and, moreover, not a stable compound under ambient conditions (we presume decomposition by oligomerization). Therefore, the crude reaction mixture was directly treated with a stoichiometric amount of di-tert-butyl dicarbonate (Boc2O), which resulted in quantitative Boc-protection of the aziridine within two hours at 25°C-30°C. Compound 9 was isolated in 73% after chromatographic purification, as material with seemingly unlimited storability; we were not able to detect any decomposition even at elevated temperature and in solution.
| S. No. | Solvent | Acid | Peracid | Time (h) | Temp (°C) | Yield (%)b |
|---|---|---|---|---|---|---|
| 1 | Dichloromethane | TFA | H2O2 | 12 | 0-30 | No product |
| Dichloromethane | AcOH | Ac2O/H2O2 | 12 | 0-30 | No product | |
| 2 | Dichloromethane | TFA | TFAA/H2O2 | 2 | <10 | 25%-30% |
| 3 | Dichloromethane | TFA | Oxone | 3-Feb | 0-30 | 20-25 |
| 4 | Dichloromethane | TFA | m-CPBA | 3-Feb | 0-30 | 85%-90% |
| 5 | Dichloromethane | Formic acid | m-CPBA | 3-Feb | 0-30 | 30-35 |
aReaction conditions: All the reaction were carried out 1-Benzyl-4-methyl-1,2,3,6-tetrahydro-pyridine (1 mmol), Trifluoroacetic acid (3.0 mmol), peracid (2.5 mmol) dichloromethane (50 ml) at room temperatures;
bIsolated yields
Table 1: Optimization of epoxydation with peracidsa
In comparison to the huge number of reports on the ring opening of aziridines by other nucleophiles [38,39], their ring opening by hydrides has received very limited interest in literature dispite the synthetic potential of this approach.The reduction of Boc aziridine 9 with LiAlH4 in THF at 0°C-10°C for 1 h, reaction continued for 5-6 h at reflux and yielded a mixture (30: 70) of ring opened methyamines derivatives (derived from hydride attack at both the more and the less hindered carbon atom in 2: 1 ratio), 10a and 10b respectively, in an overall yield of 62%.These isomers were separated by column chromatography.
After chiral resolution of compound 10a with di-para tolyltartarate, optically pure compound 11 was successfully obtained by recrystalisation in methanol-water with 43% yield. As shown in Scheme 1 the nuclephilic substitution of commertially available 4-chloro-7-tosyl-7H-pyrazolo[2,3- d]pyrimidine compound with compound 11 underwent very smoothly in shorter reaction time and an improved yeied. Without tosylprotection, nucleophilic substitution reaction was very slow and low yielded. This indicated that tosyl group effectively reduced the electron density of pyrrolopyrimidine ring by its strong electron-withdrawing and conjugative effect. Subsequently, the tosyl group was hydrolyzed in aqueous solution of sodium hydroxide to afford compound 3 in a good yield. Thus, a combination of nucleophilic substitution and hydrolysis was successfully carried out in one pot under alkaline condition.
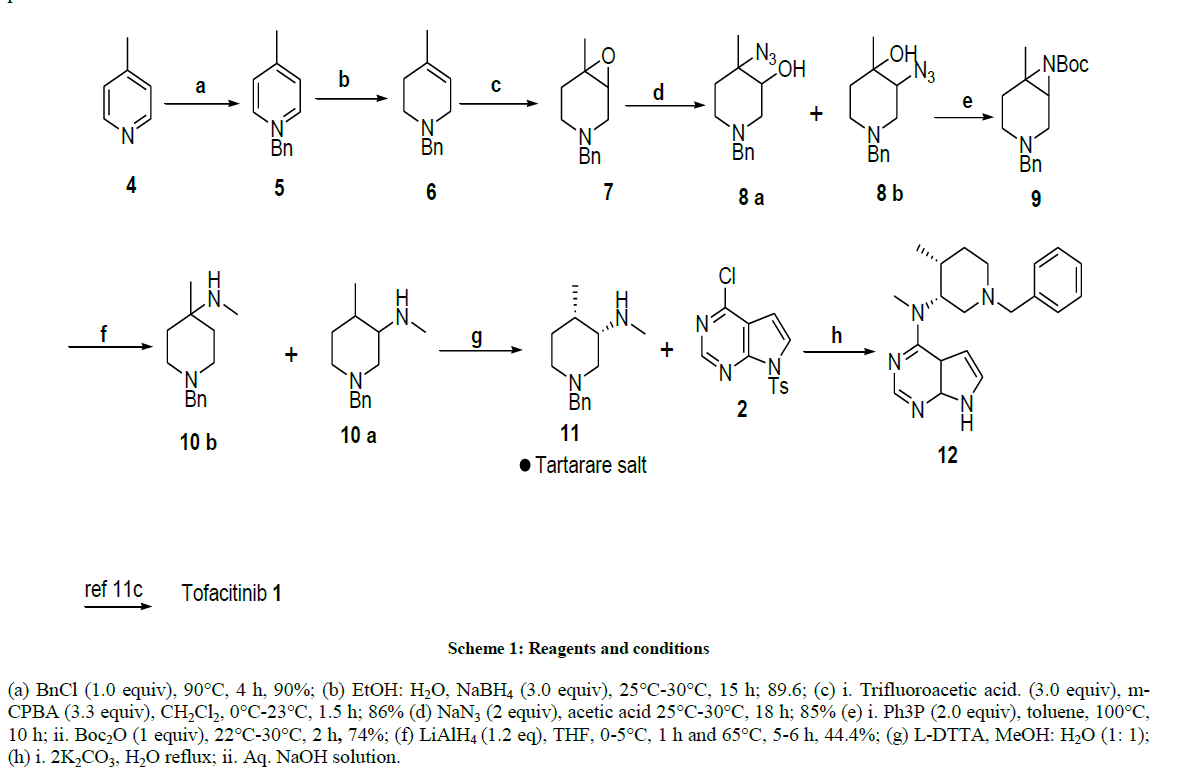
(a) BnCl (1.0 equiv), 90°C, 4 h, 90%; (b) EtOH: H2O, NaBH4 (3.0 equiv), 25°C-30°C, 15 h; 89.6; (c) i. Trifluoroacetic acid. (3.0 equiv), m- CPBA (3.3 equiv), CH2Cl2, 0°C-23°C, 1.5 h; 86% (d) NaN3 (2 equiv), acetic acid 25°C-30°C, 18 h; 85% (e) i. Ph3P (2.0 equiv), toluene, 100°C, 10 h; ii. Boc2O (1 equiv), 22°C-30°C, 2 h, 74%; (f) LiAlH4 (1.2 eq), THF, 0-5°C, 1 h and 65°C, 5-6 h, 44.4%; (g) L-DTTA, MeOH: H2O (1: 1); (h) i. 2K2CO3, H2O reflux; ii. Aq. NaOH solution.
Scheme 1: Reagents and conditions
We proposed an alternative strategy based on the precursor 3-hydroxy-4-methylpiperidine 14 (Scheme 3) which has been developed including two from Pfizer [29], and another by A. Marican et al. [26-28]. Pfizer’s route started with the benzylation of 4-pipecoline followed by reduction with sodium borohydride to afford 6, which underwent a hydroboration followed by oxidation to yield hydroxy piperidine 14 (Scheme 2), Unfortunately, the extreme reaction conditions required, and potentially hazardous nature of the reagents and releases toxic hydrogen fluoride makes these preparations unattractive for operation on a manufacturing scale and A. Marican et al. route started with chiral 5-Hydroxy-piperidin- 2-one and it was too long to industrialize and employed costly raw materials and air sensitive reagents which gave a poor product yield.
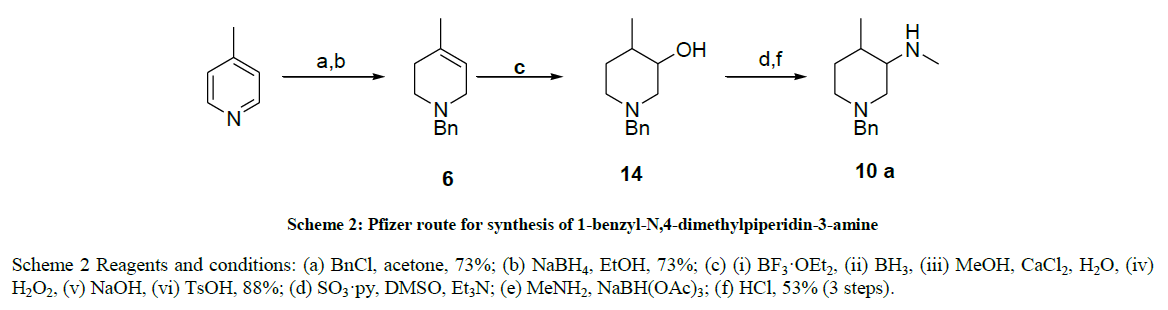
Scheme 2 Reagents and conditions: (a) BnCl, acetone, 73%; (b) NaBH4, EtOH, 73%; (c) (i) BF3·OEt2, (ii) BH3, (iii) MeOH, CaCl2, H2O, (iv) H2O2, (v) NaOH, (vi) TsOH, 88%; (d) SO3·py, DMSO, Et3N; (e) MeNH2, NaBH(OAc)3; (f) HCl, 53% (3 steps).
Scheme 2: Pfizer route for synthesis of 1-benzyl-N,4-dimethylpiperidin-3-amine
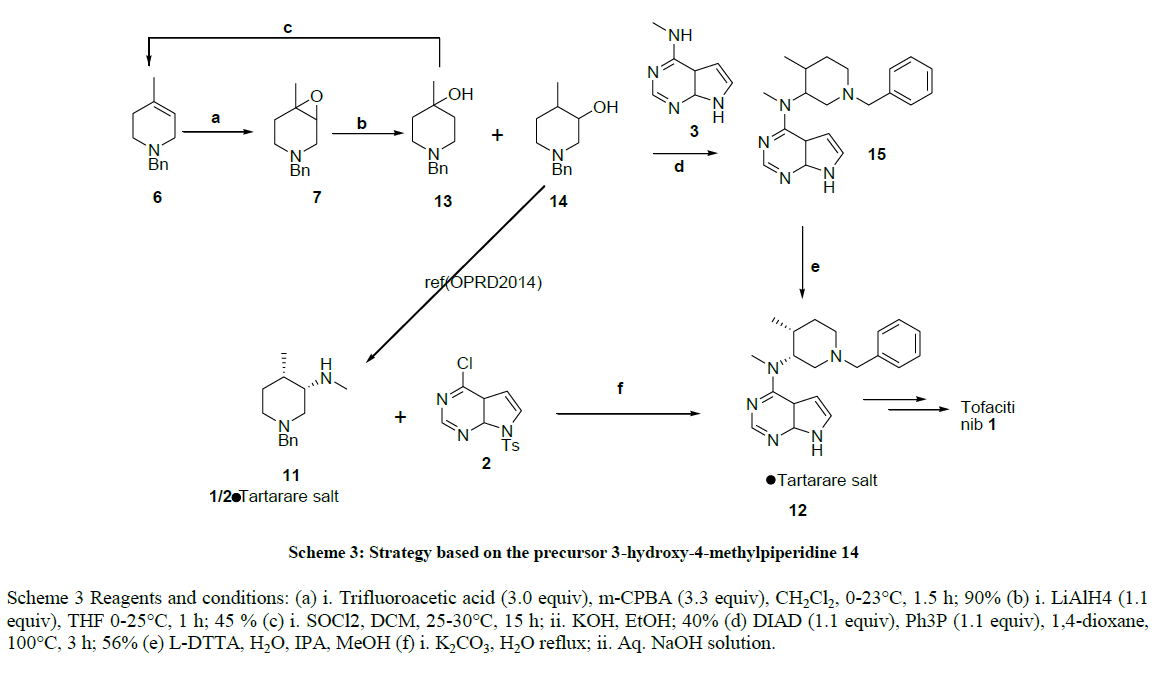
Scheme 3 Reagents and conditions: (a) i. Trifluoroacetic acid (3.0 equiv), m-CPBA (3.3 equiv), CH2Cl2, 0-23°C, 1.5 h; 90% (b) i. LiAlH4 (1.1 equiv), THF 0-25°C, 1 h; 45 % (c) i. SOCl2, DCM, 25-30°C, 15 h; ii. KOH, EtOH; 40% (d) DIAD (1.1 equiv), Ph3P (1.1 equiv), 1,4-dioxane, 100°C, 3 h; 56% (e) L-DTTA, H2O, IPA, MeOH (f) i. K2CO3, H2O reflux; ii. Aq. NaOH solution.
Scheme 3: Strategy based on the precursor 3-hydroxy-4-methylpiperidine 14
According to our retrosynthetic analysis applied to the target CP-690,550 (Figure 1), compound 14 was prepared via selective reduction of epoxide compound 7, after several parameters were evaluated in an effort to optimize the selectivity and reproducibility of the reduction of epoxide 7 with different reducing agent as shown in Table 2. However, we found that sodium borohydride and other reducing agents were not suitable for this conversion.Lithium aluminum hydride was chosen as the reducing agent as it afforded a high yield of compound 14 and the reaction was simple to perform and work up. Finally compound 14 was obtained with Lithium Aluminium Hydride (LiAlH4) in THF at 0-5°C for 1 h, the regeoisomer 13 and 14 was separated by column chromatography with 40-45% yield, And the compound 13 converted to compound 6 with 45% yield as described in the literature [31].
| S. No. | Solvent | Reagent | Temp (°C) | Time (h) | Yield (%) | |
|---|---|---|---|---|---|---|
| 13 | 14 | |||||
| 1 | THF | NaCNBH4 | 25-30 | 12 | - | - |
| 2 | THF | NaBH4 | 25-30 | 12 | - | - |
| 3 | THF | LiAlH4 | 0-5 | 1 | 40 | 41 |
| 4 | THF | LiAlH4 | 25-30 | <0.5 | 45 | 40 |
| 5 | Ether | LiAlH4 | 0-5 | 1 | 42 | 45 |
aReaction conditions: All the reactions were carried out 3-Benzyl-6-methyl-7-oxa-3-aza-bicyclo[4.1.0]heptanes (1.0 mmol.), Reducing agent(1.1 mmol.), and THF (10 ml) at room temperatures;
bIsolated yields
Table 2: Optimization of reducing agents for the synthesis of 1-Benzyl-4-methyl-piperidin-3-olza
The reaction between 14 and commercially available Methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine proceeded under Mistsunobu conditions similar to those described in the literature [26-28]. But required a shorter time and gave an acceptable yield of advanced tofacitinib intermediate 15, which was resolved with di-para tolyltartarate as described in the literature [40].
Conclusion
We have developed an alternate route for the synthesis of 10a and 14, which are important intermediate for the synthesis of tofacitinib from N-benzyl 3,4-epoxypiperidine, this synthetic strategy provides reliable new entries for the synthesis of tofacitinib, as well as synthesis of piperidine based bioactive compounds. The key reaction was the reductive ring opening of Boc activated aziridine as well as epoxide utilizing LiAlH4 has been reported for the first time in high regioselective way and it allows to exclude an operation with a toxic hydrogenfluoride and oxidation to make key intermediate of tofacitanib.
References
- D.A. Horton, G.T. Bourne, M.L. Smythe, Chem. Rev., 2003, 103, 893.
- S. Källström, R. Leino, Bioorg. Med. Chem., 2008, 16, 601.
- C. De Risi, G. Fanton, G.P Pollini, C. Trapella, F. Valente, V. Zanirato, Tetrahedron: Asymmetry., 2008, 19, 131.
- S. Laschat, T. Dickner, Synthesis., 2000, 1781.
- E. Vilsmaier, J. Prakt. Chem., 2000, 342, 218.
- C. Escolano, M Amat, J. Bosch, Chem. Eur. J., 2006, 12, 8198.
- M. Eckhardt, N. Hauel, F. Himmelsbach, E. Langkopf, H. Nar, M. Mark, M Tadayyon, L. Thomas, B. Guth, R. Lotz, Bioorg. Med. Chem. Lett., 2008, 18, 3158.
- M. Eckhardt, N. Hauel, E. Langkopf, F. Himmelsbach, Tetrahedron Lett., 2008, 49, 1931.
- J. Jin, Y. Wang, D. Shi, F. Wang, R.S. Davis, Q. Jin, W. Fu, J.J. Foley, E.F. Webb, C.J. Dehaas, M. Berlanga, M. Burman, H.M. Sarau, D.M. Morrow, P. Rao, L.A. Kallal, M.L. Moore, R.A. Rivero, Palovich, Queen's University, Copyrighted material, 1662 H, Schramm et al. PAPER Synthesis., 2009, 10, 1659-1662 © Thieme Stuttgart, New York, M. Salmon, M. Belmonte, K.E. Busch-Petersen, J. Med. Chem., 2008, 51, 4866.
- J.L. Diaz, D. Fernandez-Forner, J. Bach, R. Lavilla, Synth. Commun., 2008, 38, 2799.
- E. Martini, C. Ghelardini, S. Dei, L. Guandalini, D. Manetti, M. Melchiorre, M. Norcini, S. Scapecchi, E. Teodori, M.N. Romanelli, Bioorg. Med. Chem., 2008, 16, 1431.
- S.D. Erickson, B. Banner, S. Berthel, K. CondeKnape, F. Falcioni, I. Hakimi, B. Hennessy, R.F. Kester, K. Kim, C. Ma, W. McComas, F. Mennona, S. Mischke, L. Orzechowski, Y. Qian, H. Salari, J. Tengi, K. Thakkar, R. Taub, J.W. Tilley, H. Wang, Bioorg. Med. Chem. Lett., 2008, 18, 1402.
- B.A. Acker, E.J. Jacobsen, B.N. Rogers, D.G. Wishka, S.C. Reitz, D.W. Piotrowski, J.K. Myers, M.L. Wolfe, V.E. Groppi, B.A. Thornburgh, P.M. Tinholt, R.R. Walters, B.A. Olson, L. Fitzgerald, B.A. Staton, T.J. Raub, M. Krause, K.S. Li, W.E. Hoffmann, M. Hajos, R.S. Hurst, D.P. Walker, Bioorg. Med. Chem. Lett., 2008, 18, 3611.
- J.J.W. Duan, L. Chen, Z. Lu, C.B. Xue, R.Q. Liu, M.B. Covington, M. Qian, Z.R. Wasserman, K. Vaddi, D.D. Christ, J.M. Trzaskos, R.C. Newton, C.P. Decicco, Bioorg. Med. Chem. Lett., 2008, 18, 241.
- Y. Zhou, C. Chow, D.E. Murphy, Z. Sun, T. Bertolini, J.M. Froelich, S.E. Webber, T. Hermann, D. Wall, Bioorg. Med. Chem. Lett., 2008, 18, 3369.
- D.E. Levy, D.X. Wang, Q. Lu, Z. Chen, J. Perumattam, Y.J. Xu, J. Higaki, H. Dong, A. Liclican, M. Laney, B. Mavunkel, S. Dugar, Bioorg. Med. Chem. Lett., 2008, 18, 2395.
- R.C.A. Isaacs, M.G. Solinsky, K.J. Cutrona, C.L. Newton, A.M. Naylor-Olsen, D.R. McMasters, J.A. Krueger, S.D. Lewis, B.J. Lucas, L.C. Kuo, Y. Yan, J.J. Lynche, E.A. Lyle, Bioorg. Med. Chem. Lett., 2008, 18, 2062.
- M. Mishra, R. Kolhatkar, J. Zhen, I. Parrington, M.E.A. Reith, A.K. Dutta, Bioorg. Med. Chem., 2008, 16, 2769.
- X.E. Hu, Tetrahedron Lett., 2002, 43, 5315.
- M.W. Majchrzak, A. Kotelko, J.B. Lambert, Org. Magn. Reson., 1983, 21, 539.
- J.J. Kulagowski, W. Blair, R.J. Bull, C. Chang, G. Deshmukh, H.J. Dyke, C. Eigenbrot, N. Ghilardi, P. Gibbons, T.K. Harrison, P.R. Hewitt, M. Liimatta, C.A. Hurley, A. Johnson, T. Johnson, J.R. Kenny, P. Bir Kohli, R.J. Maxey, R. Mendonca, K. Mortara, J. Murray, R. Narukulla, S. Shia, M. Steffek, S. Ubhayakar, M. Ultsch, A. Van Abbema, S.I. Ward, B. Waszkowycz, M. Zak, J. Med. Chem., 2012, 55, 5901-5921.
- J.K. Jiang, K. Ghoreschi, F. Deflorian, Z. Chen, M. Perreira, M. Pesu, J. Smith, D.T. Nguyen, E.H. Liu, W. Leister, S. Costanzi, J.J. O’Shea, C.J. Thomas, J. Med. Chem., 2008, 51, 8012-8018.
- L. Vijayakrishnan, R. Venkataramana, P. Gulati, Trends Pharmacol. Sci., 2011, 32, 25-34.
- M.M. Seavey, P. Dobrzanski, Biochem. Pharmacol., 2012, 83, 1136-1145.
- G.V. Grishina, A.A. Borisenko, I.S. Veselov, A.M. Petrenko, Russian J. Org. Chem., 2005, 41(2), 272-278.
- M. Adolfo, J.S. Mario, S.S. Leonard, Tetrahedron Lett., 2013, 10, 5096-5098.
- B.Y., J.Q. Liu, W.H. Zhang, X.Z. Chen, Synthesis., 2011, 8, 1208-1212.
- Preparation method of tofacitinib, CN 103819474 A, 2014.
- D.H. Brown Ripin, S. Abele, W. Cai, T. Blumenkopf, J.M. Casavant, L. Doty, M. Flanagan, C. Koecher, K.W. Laue, K. McCarthy, C. Meltz, M. Munchhoff, K. Pouwer, B. Shah, J. Sun, J. Teixeira, T. Vries, D.A. Whipple, G. Wilcox, Org. Process Res. Dev., 2003, 7, 115-120.
- W. Cai, J.L. Colony, H. Frost, J.P. Hudspeth, P.M. Kendall, A.M. Krishnan, T. Makowski, D.J. Mazur, J. Phillips, D.H. Brown Ripin, S.G. Ruggeri, J.F. Stearns, White, Y.S. Patil, N.L. Bonde, A.S. Kekan, D.G. Sathe, A. Das, Org. Process Res. Dev., 2014, 18, 1714-1720.
- M.A. Iorio, P. Ciuffa, G. Damia, Tetrahedron., 1970, 26(23), 5519-5527.
- K.C. Kumara Swamy, N.N. Bhuvan Kumar, E. Balaraman, K.V.P. Pavan Kumar, Chem. Rev., 2009, 109, 2551.
- S. Fletcher, Org. Chem. Front., 2015, 2, 739.
- S.D Larsen, P.A.J. Grieco, Am.Chem.Soc., 1985, 107, 1768-1769.
- D.H. Brown Ripin, S. Abele, W. Cai, T. Blumenkopf, J.M. Casavant, J.L. Doty, M. Flanagan, C. Koecher, K.W. Laue, K. McCarthy, C. Meltz, M. Munchhoff, K. Pouwer, B. Shah, J. Sun, J. Teixeira, T. Vries, D.A. Whipple, G. Wilcox, Org. Process Res. Dev., 2003, 7, 115.
- J. Christoffers, Y. Schulze, J. Pickardt, Tetrahedron., 2001, 57, 1765.
- J.J. Baldwin, D.A. Claremon, C. Tice, S. Cacatian, L.W. Dillard, A.V. Ishchenko, J. Yuan, Z. Xu, G. McGeehan, R.D. Simpson, S.B. Singh, W. Zhao, P.T. Flaherty, WO 2007/070201 A1, Chem. Abstr., 2007, 147, 95545.
- X.E. Hu, Tetrahedron., 2004, 60, 2701-2743.
- P. Lu, Tetrahedron., 2010, 66, 2549-2560.
- Efficient method for the preparation of tofacitinib citrate, EP 3078665 A1, 2012.
Supporting Information
General Information
All reagents were used as received from commercial sources without further purification or prepared as described in the literature. Reactions were stirred using Teflon-coated magnetic stirring bars. TLC plates were visualized by ultraviolet light. Chromatographic purification of products was carried out by flash column chromatography on silica gel (100-200 mesh). Melting points were determined using an electro thermal melting point apparatus and are uncorrected. NMR spectra were measured in CDCl3 and DMSO-d6 (all with TMS as internal standard) on a Varian Gemini 400 MHz FT NMR spectrometer magnetic resonance spectrometer. Chemical shifts (delta) are reported in ppm, and coupling constants (J) are in Hz. The following abbreviations were used to explain the multiplicities: s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet. LCMS were recorded on an Agilent HP-5989A quadrapole mass spectrometer.
Experimental Procedures and Analytical data
Synthesis of benzyl pyridinium chloride (5)
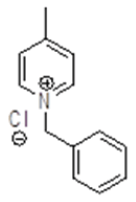
To a stirring solution of 4-methyl Pyridine 4 (25.0 g, 268.81 mmol, 1.0 eq.) cooled to 0-5°C and benzyl chloride (36.96 g, 261.81 mmol, 1.0 eq.) was added slowly drop wise, after complete addition, stirring was continued at 90°C for 3-4 h. After completion of reaction on TLC, reaction mass brought to rt, stirred for 1 hour, filtered the precipitated white crystalline solid (53.0 g, 242.00 mmol, 90.0%).
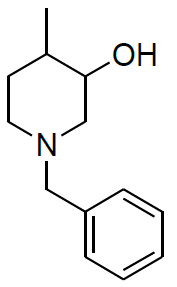
Synthesis of 1-benzyl-1,2,3,6-tetrahydro-4-methylpyridineride(6)
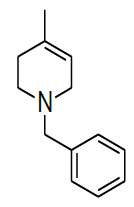
Procedure: To a stirring solution of benzyl pyridinium chloride 5 (30.0 g, 136.98 mmol, 1.0 eq.) in a EtOH: H20 (9: 1) (300 ml) cooled to 0-10° C and NaBH4 (15.61 g, 410.95 mmol, 3.0 eq.) was added Portion wise, after complete addition, stirring was continued at RT for 16 h. reaction was monitored by TLC, After completion of reaction diluted with water and extracted with Ethyl acetate (2 × 250 ml), combined organic layer was washed with brine solution, dried over sodium sulphate, the solvent was evaporated under reduced pressure to get desired compound as pale yellow liquid which was purified by column chromatography using 100-200 silica mesh with 10% ethyl acetate in hexane (23.0 g, 122.90 mmol, 89.9%).
For compound 6: 1H-NMR (400 MHz, CDCl3) δ (ppm)=7.35-7.33 (m, 5H), 5.36-5.34 (m, 1H), 3.56 (s, 3H), 2.92 (t, J=3.2 Hz, 2H), 2.54 (t, J=5.6 Hz, 2H), 2.06 (d, J=7.6 Hz, 2H), 1.67 (s, 3H). LC-MS: tRet=0.608 min, M+1: 188.2.
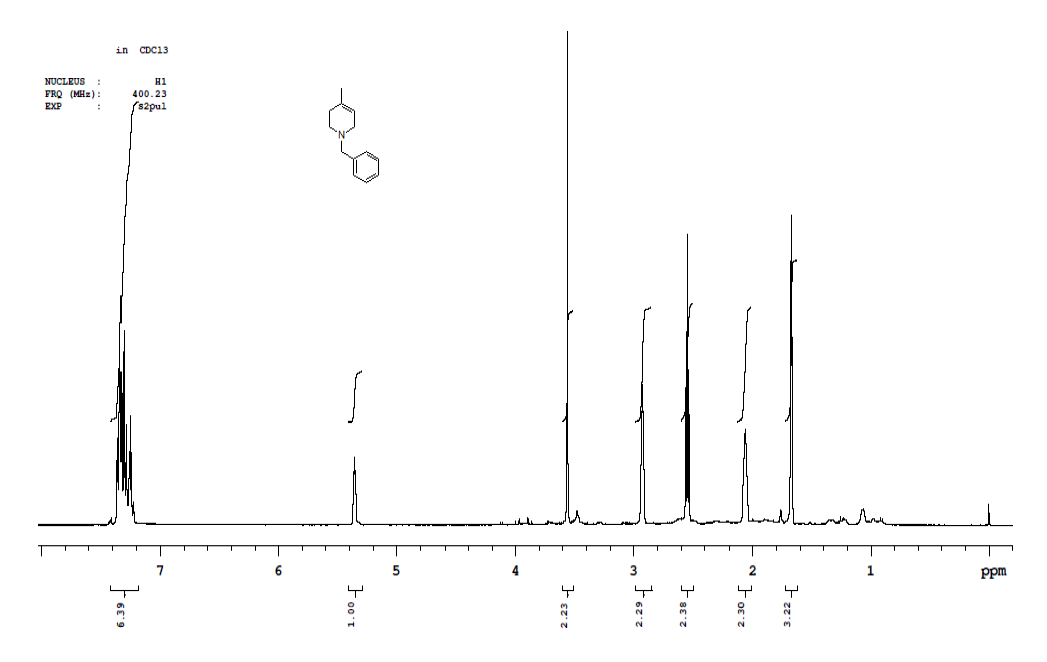
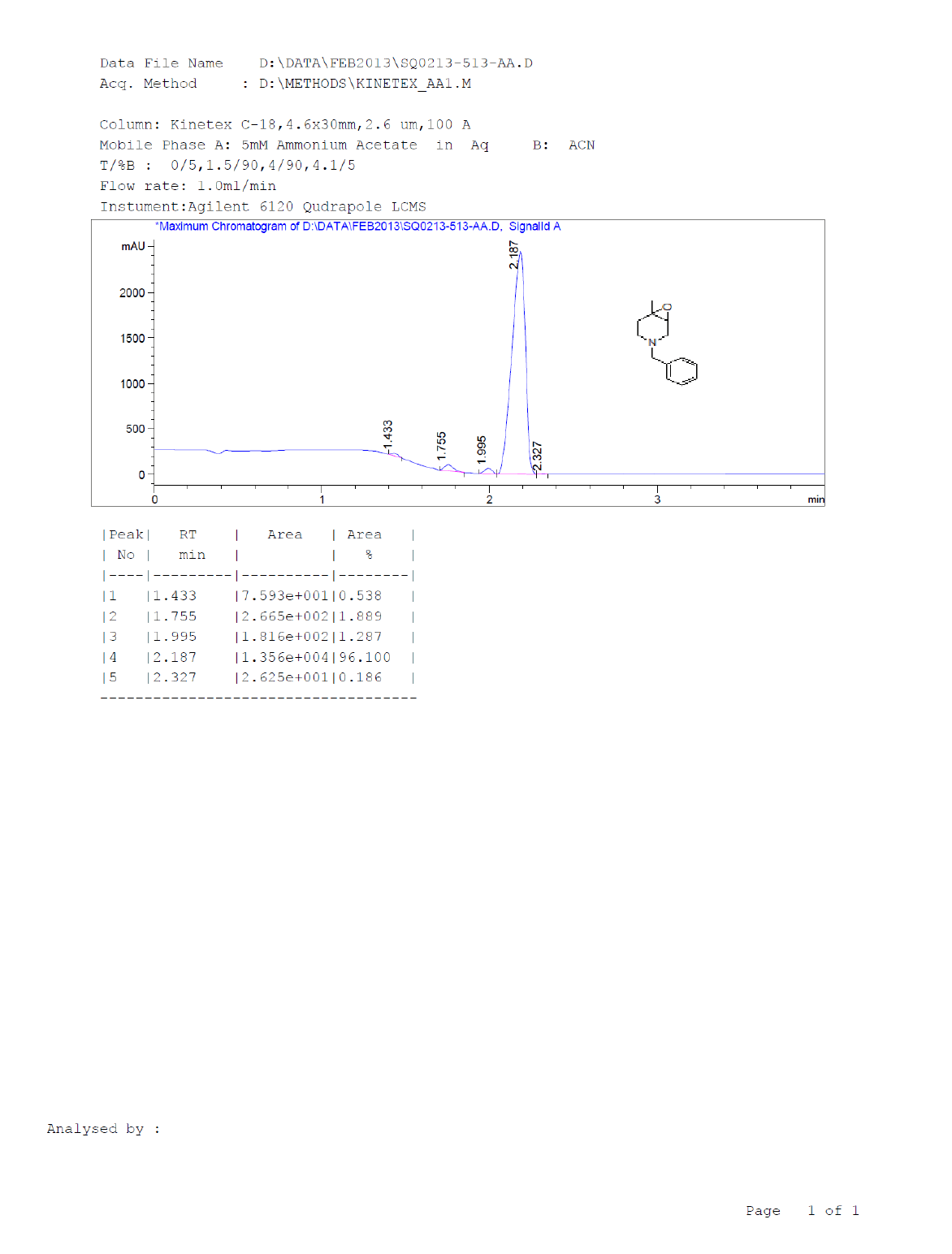
Synthesis of 4-benzyl-1-methyl-7-oxa-4-aza-bicyclo [4.1.0] heptane (7)
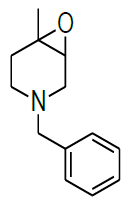
Procedure: To stirred solution of 1-benzyl-1,2,3,4-tetrahydro-4-methylpyridine (30.0 g, 160.42 mmol), in dichloromethane, was added trifluoroacetic acid (30.89 ml, 401.06, 2.5 eq) at 05°C and stirred for 30 min same temparature and added m-CPBA (82.8 g, 481.28 mmol, 3.0 eq, 77% purity)portion wise at same temparature and reaction continued for 2-3 h at 25-30°C.Reaction progress was monitired by TLC, after completion of reaction, diluted with cooled water, excess m-CPBA quenched with saturated sodium sulfite, Organic layer was separated and washed with sat. NaHCO3 followed by brine solution, dried over sodium sulphate, filtered the solvent, evaporated under reduced pressure to get crude compound which was purified by column chromatography using 100-200 silica mesh with 10% ethyl acetate in hexane as a colorless liquid. (28.2 g, 203.50 mmol, 86%).
Compound 7 : 1H-NMR (400 MHz, CDCl3) δ (ppm)=7.3-7.24 (m, 5H), 3.04 (d, J=4.1 Hz, 1H), 3.01 (d J=9.2 Hz, 1H), 2.41 (d, J=12.8 Hz, 1H), 2.32 (d, J=10.0 Hz, 1H), 2.01 (s(broad), 1H), 1.83(d, J=10.0 Hz, 2H), 1.26 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ (ppm)=20.25, 52.57, 57.85, 62.29, 68.25, 122.58, 127.11, 128.21, 128.90, 134.97, 137.82. LC-MS: tRet=2.187; min, M+1: 204.2.
Synthesis of trans 4-azido-1-benzyl-4-methylpiperidin-3-ol (8a) and trsns 3-azido-1-benzyl-4-methylpiperidin-4-ol (8b)
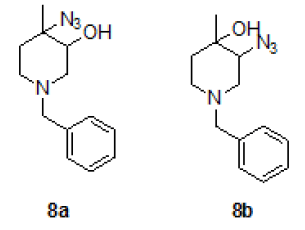
To a stirred solution of mixture of epoxide 8 (25.0 g, 122.54 mmol, 1.0 eqt.), in water acetic acid (6: 4) 250 ml, to this NaNO3 (22.5 g, 367.62 mmol 3.0 eqt.) was added at RT and stirred for overnight at same temperature. Reaction was monitored by TLC. After completion of reaction, neutralized with Sodium bicarbonate solution, and extracted with ethyl acetate (2 × 200 ml), combined organic layer was washed with brine solution, dried over sodium sulphate, the solvent was evaporated under reduced pressure and the residue was purified by column chromatography using 100-200 silica mesh with ethyl acetate in hexane to give a mixture of both regioisomers (11a/11b = 96: 04 by LCMS) as a pale yellow liquid. The isomers could be separated by repeated chromatography. Compound 8a was eluted at 20% EtOAc in hexane (18.2 g, 73.68 mmol, 60.6%) and compound 8b was eluted 30% EtOAc in hexane (2.4 g, 9.716 mmol, 8.0%).
For compound 8a: 1H-NMR (400 MHz, CDCl3): 7.33-7.34 (m, 5H), 3.52 (s, 2H), 3.36(s, 1H), 2.66-2.56 (m, 3H), 2.32-2.26 (m, 1H), 2.87-2.81 (m, 1H), 1.60-1.57 (m, 1H), 1.38 (s, 3H). LC-MS: tRet=1.62 min, M+1: 247.2.
Synthesis of tert-butyl 4-benzyl-1-methyl-4,7-diaza-icyclo[4.1.0]heptane-7-carboxylate(9)
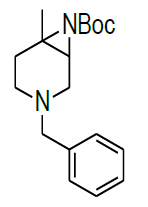
A mixture of both regioisomers of azide 8a/8b (20.00 g, 82.3 mmol 1.0 eqt.), PPh3 (62.0 g, 164.50 mmol 2.0 eqt.), and toluene (200 ml) was stirred for 12 h at 100°C under an N2-atmosphere.Reaction was monitored by TLC. The mixture was cooled to 25-30 °C and a solution of Boc2O (26.9 g, 123.45 mmol 1.5 eqt), was added. After stirring the mixture for 2 h at 25-30°C, the solvent was removed under vacuum and the residue was purified by column chromatography using 100-200 silica mesh with 20% ethyl acetate in hexane as a colorless liquid, (18.0 g, 59.40 mmol, 74%).
For Compound 9: 1H-NMR (400 MHz, CDCl3): 7.32-7.23 (m, 5H), 3.40 (s, 2H), 2.96-2.92 (m, 1H), 2.50-2.42 (m, 1H), 2.25-2.22 (m, 1H), 2.05- 2.03 (m, 1H), 1.86-1.82 (m, 1H), 1.69-1.64 (m, 1H), 1.38 (s, 9H), 1.22 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ (ppm)=25.20, 25.5, 35.4, 37.7, 45.5, 46.5, 57.85, 62.29, 78.25, 127.11, 128.21, 128.90, 134.97, 137.82. LC-MS: tRet=1.75 min, M+1: 303.2.
Synthesis of 1-benzyl-N,4-dimethylpiperidin-3-amine (10a)
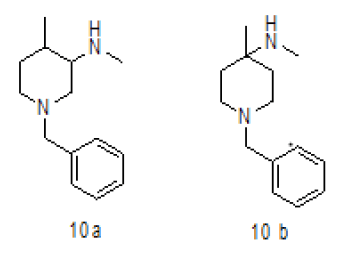
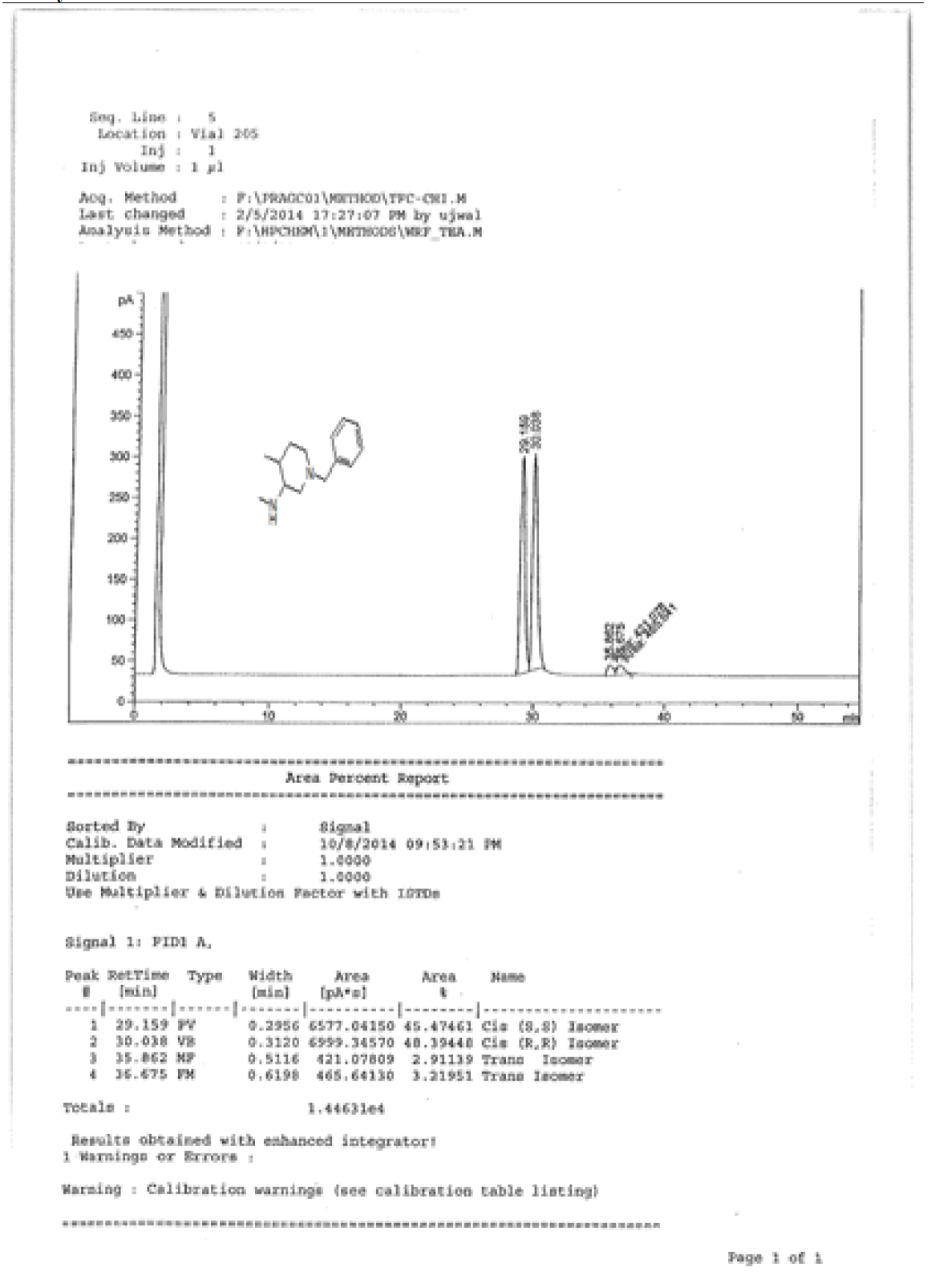
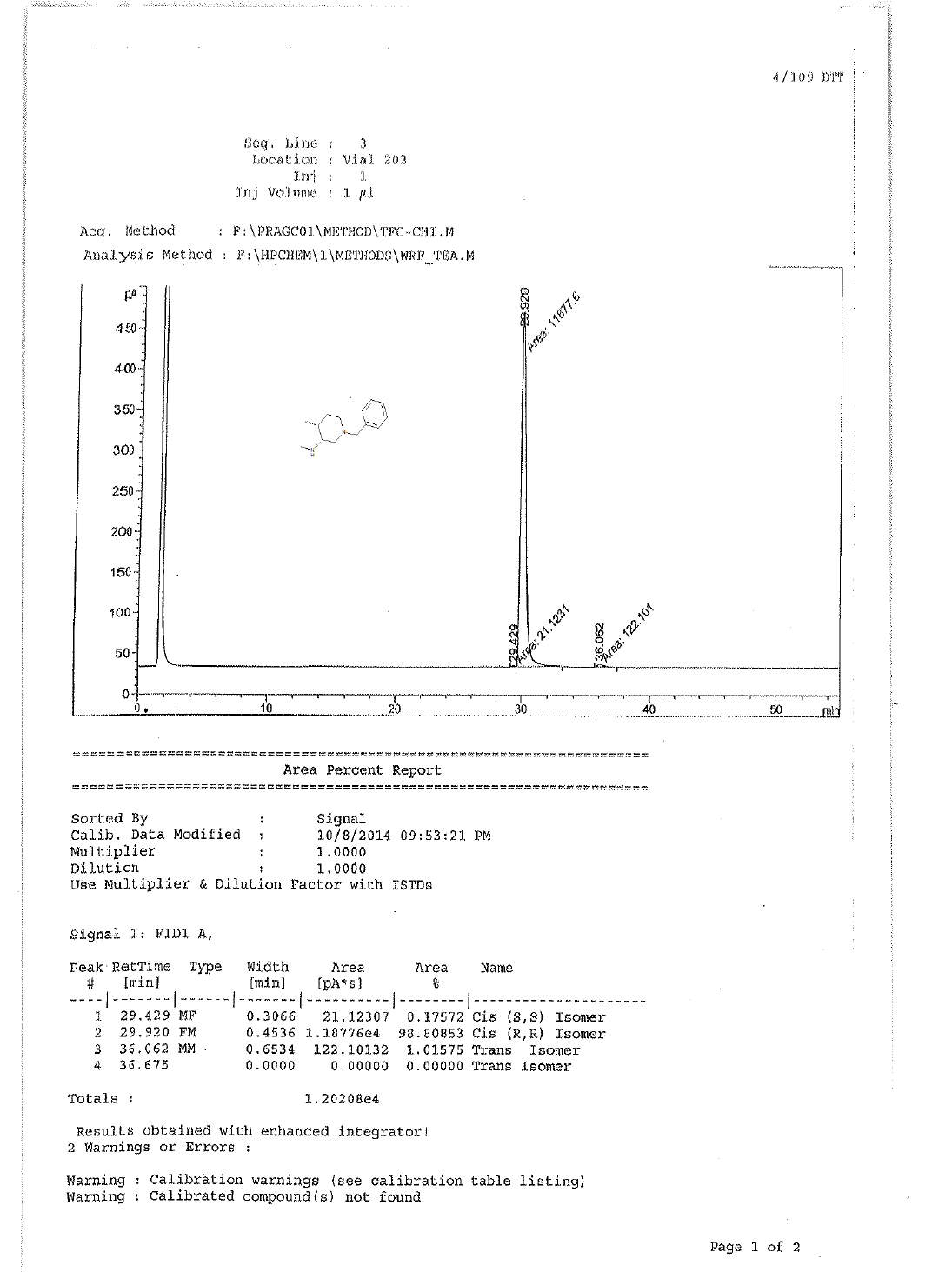
To a suspension LiAlH4 (3.76 g, 99.00 mmol) in THF (100.0 ml) was added 9 (10.0 g, 33.00 mmol) in THF (20.0 ml) at 0-5°C.The mixture was stirred for 1 h at room temperature and then refluxed for 4-5. After completion of reaction monitored by TLC, reaction mass brought to 0-5°C, the mixture was quenched with ethyl acetate followed by sat.Na2SO4 and filtered. The filtrate was diluted with water (100 ml) and extracted with EtOAc (3 × 100 ml). The combined organic layers were washed with brine and dried over Na2SO4. The solvent was filtered and concentrated under reduced pressure to get crude regioisomers, which were purified by column chromatography using 100-200 silica mesh using Methanol in dichloromethane to give desired products 10a as colorless oil (3.2 g, 19.26 mmol, 44.4%) and 10b as colorless liquid (1.2 g, 16.6%), compound 10 a was directly used in chiral resolution.
For compound 10a: 1H-NMR (400 MHz, CDCl3) δ (ppm)=7.20-7.28 (m, 5H), 5.25 (d, J= 8.8 Hz, 1H), 3.70 (d, J= 9.2 Hz, 1H), 3.52-3.60 (m, 1H), 3.43 (ABq, J=13.2 Hz, 2H), 2.76 (d, J=10.4, 1H), 2.12 (d, J=10.8 Hz, 1H), 1.90 (td, J=11.2, 3.2 Hz, 1H), 1.48-1.64 (m, 1H), 1.42 (s, 9H), 1.25-1.40 (m, 2H), 0.90 (d, J=8.4 Hz, 3H). LC-MS: tRet=1.75 min, M+1: 218.34.
Synthesis of (3S, 4S)-1-benzyl-N-4-dimethylpiperidine-3-amine. Tartarate salt (11)
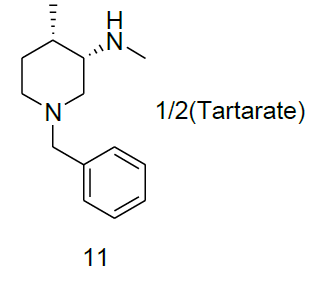
Compound 10a (3.2 g, 14.67 mmol) was added in the mixture of methanol (5.0 ml) and isopropyl alcohol (20.0 ml), followed by the addition of water(20.0 ml) and di-p-toluoyl-L-tartarate (2.8 g, 7.33 mmol). The reaction mixture was heated to reflux until homogeneous. At this temperature reaction stirred for 1 h, then slowly reaction brought to rt and filtered the target compound 11 (2.2 g, mmol, 50.2%) as white solid. For compound 13a: 1H-NMR (400 MHz, CD3OD) δ (ppm)=8.04 (d, J=8.0 Hz, d), 7.20-7.28 (m, 7H), 5.85 (s, 1H), 3.70 (d, J= 12.7 Hz, 1H), 3.42 (d, J=12.7 Hz, 1H), 3.09 (s, 1H), 2.91 (d, J=23.4 Hz, 2H), 2.49 (s, 1H),2.22 (d, J= 18.3, 2H), 1.91 (q, J=3.4 Hz, 1H), 1.47-1.62 (m, 2H), 1.02 (d, J=7.1 Hz, 3H).
Synthesis of N-((3R,4R)-1-benzyl-4-methylpiperidin-3-yl)-7,7a-dihydro-N-methyl-4aH-pyrrolo[2,3-d]pyrimidin-4-amine (12)
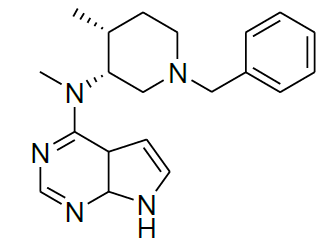
To a stirred solution of 11 (2.0 g, 3.31 mmol), in 20 ml of water, was added potassium carbonate (1.37 g, 9.93 mmol), and 4-chloro-7-tosyl-7H-pyrrolo[2,3- d]pyrimidine 2 (2.04 g, 6.62 mmol). The reaction mixture was stirred at reflux for 10 h. After completion of reaction monitored by TLC. Then, the reaction mixture was cooled to room temperature, saturated NaOH solution (25 ml) was added at below 30°C. The resulting reaction mixture was refluxed for 6 h. After being cooled to room temperature, the reaction mixture was stirred for 2 h at room temperature. The solid was isolated by filtration to afford off-white solid 12 (0.9 g, 81.0%).
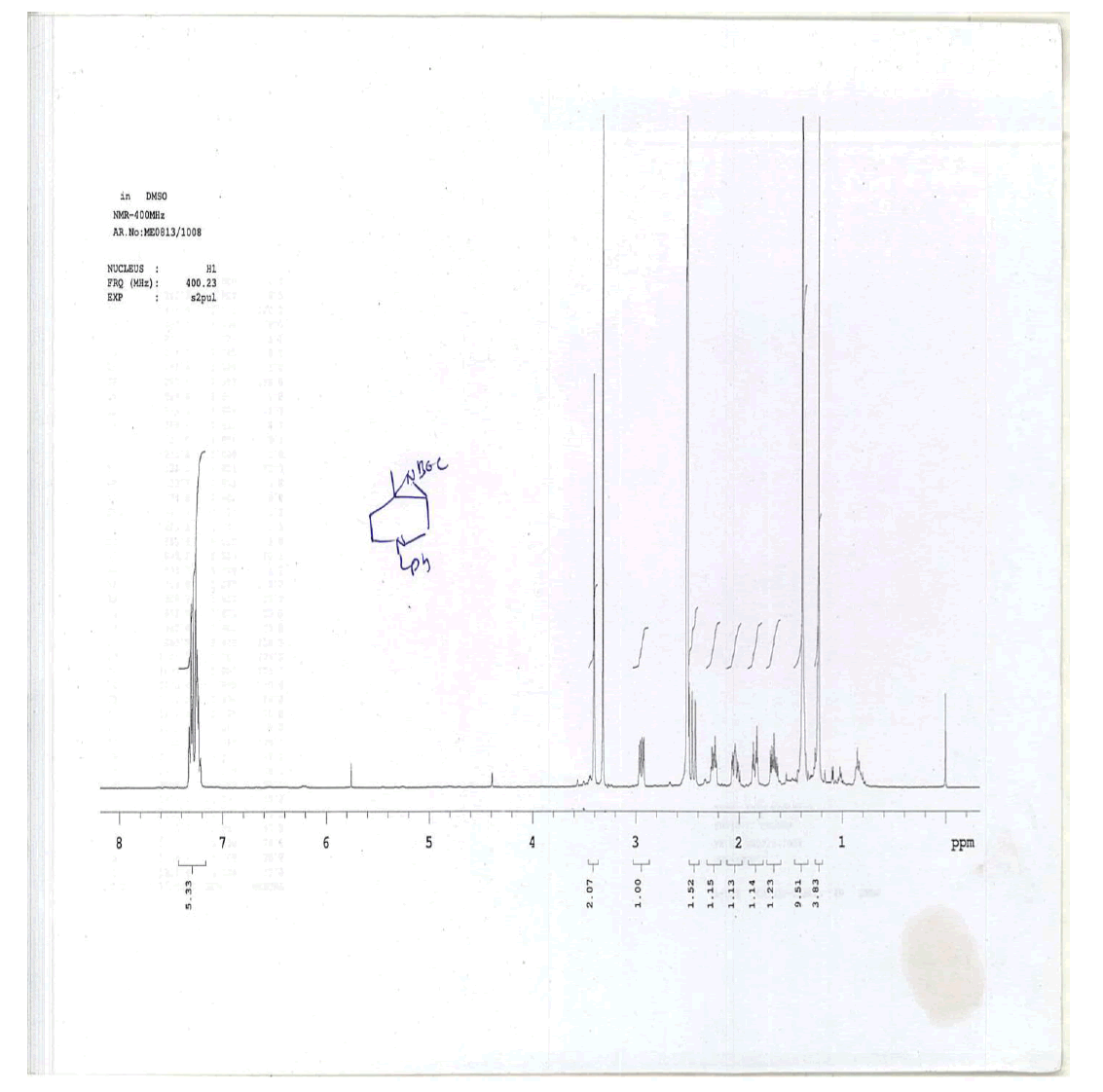
For compound 12: [α]25=+29.4 (c 1.01, MeOH); purity: 99.8% (C18 HPLC); 100% (chiralpak ID, 95:5 hexane/ isopropanol, retention time of 16.86 min); 1H-NMR (400 MHz, DMSO-d6) δ (ppm)=0.88 (d, J=6.8 Hz, 3H), 1.59-1.63 (m, 1H), 1.70 (s, 1H), 2.13 (s, 1H), 2.28 (s, 1H), 2.54 (dd, J1=4.0 Hz, J2=11.2 Hz, 1H),2.61 (s, 1H), 2.77 (dd, J1=6.0 Hz, J2=11.2 Hz, 1H), 3.44-3.52(m, 5H), 5.09 (s, 1H), 6.53 (s, 1H), 7.08 (t, J=2.6 Hz, 1H),7.20-7.23 (m, 1H), 7.28-7.31 (m, 4H), 8.05 (s, 1H), 11.55 (s,1H).
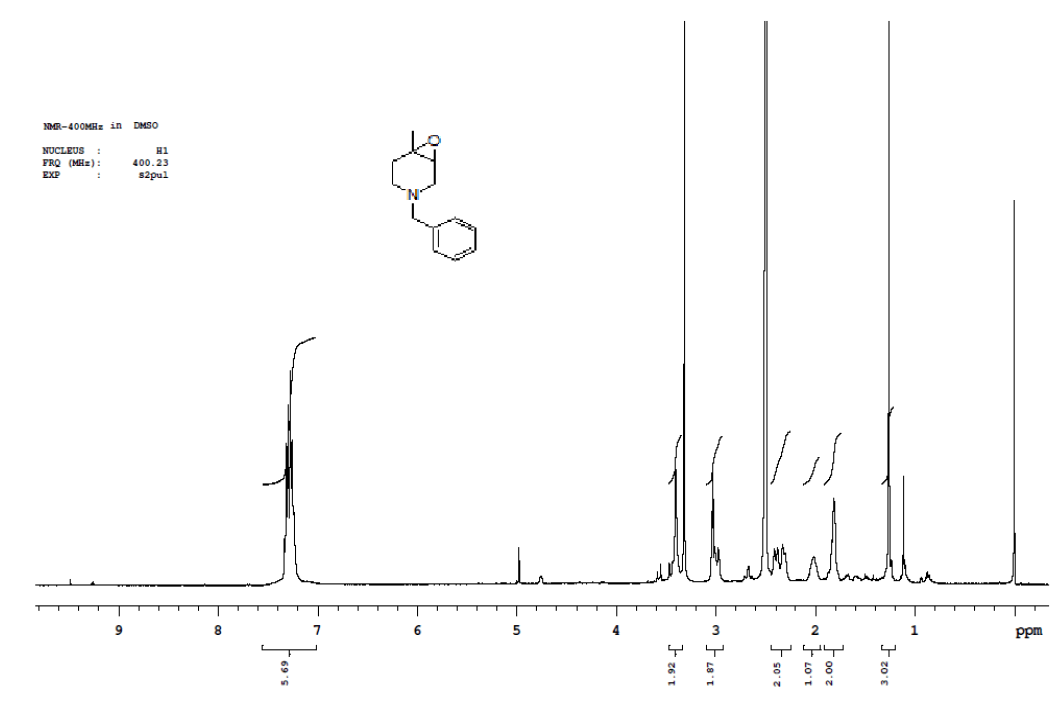
Synthesis of 1-benzyl-4-methylpiperidin-3-ol (14)
To a suspension LiAlH4 (1.87 g, 49.261 mmol) in THF (100.0 ml) was added 7 (10.0 g, 49.261 mmol) in THF (20.0 ml) at 0-5°C.The mixture was stirred for 1 h at same temperature.After completion of reaction monitored by TLC, the mixture was quenched with ethyl acetate followed by sat.Na2SO4 paste at same temperature and filtered. The filtrate was diluted with water (100 ml) and extracted with EtOAc (3 × 100 ml). The combined organic layers were washed with brine and dried over Na2SO4. The solvent was filtered and concentrated under reduced pressure to get crude regioisomers, which were purified by column chromatography using 100-200 silica mesh using ethyl acetate and hexane to give desired products 14a as colorless oil (3.8 g, 18.536 mmol, 37.6%) and 10b as colorless liquid (4.1 g, 20.00 mmol, 40.6).
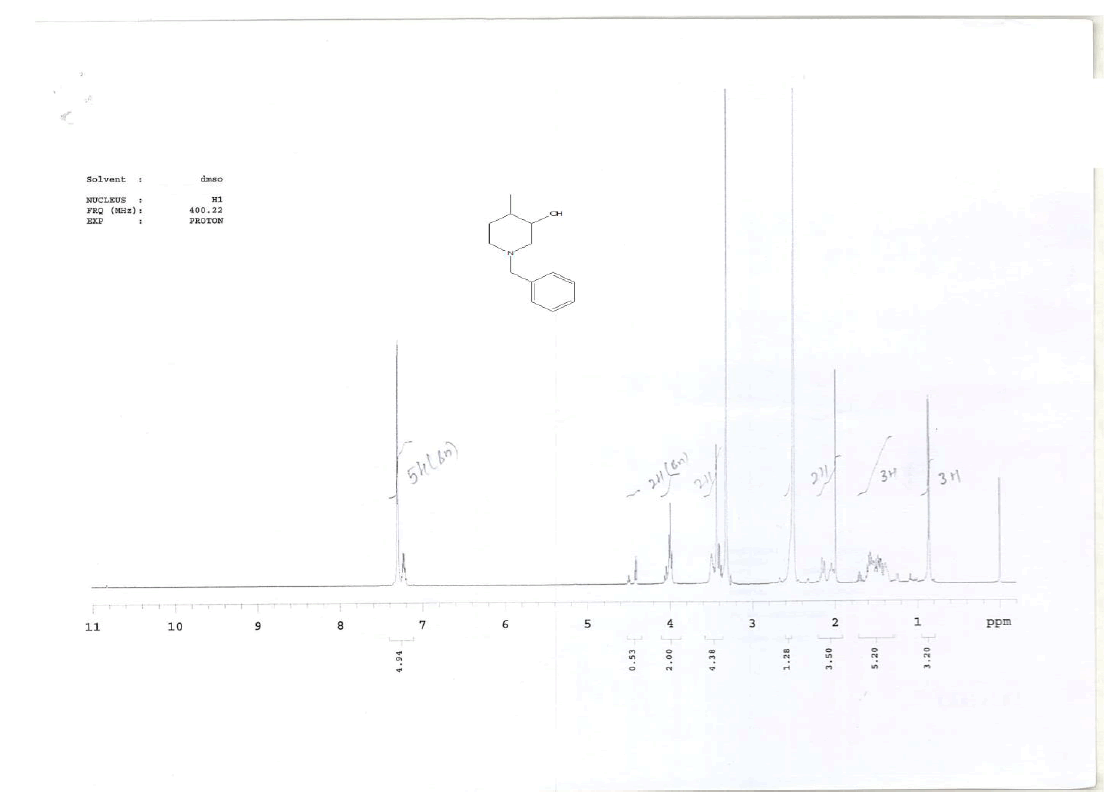
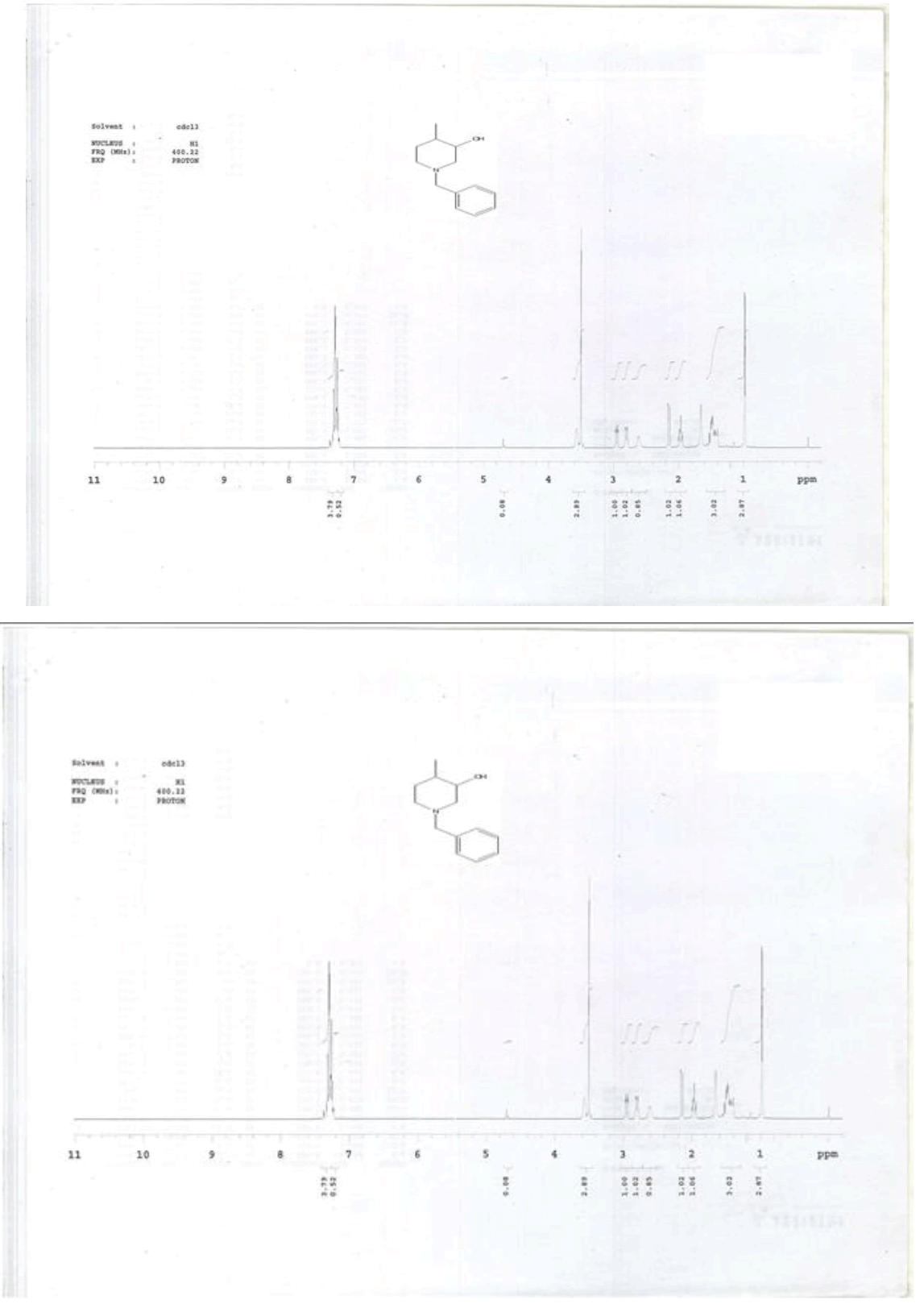
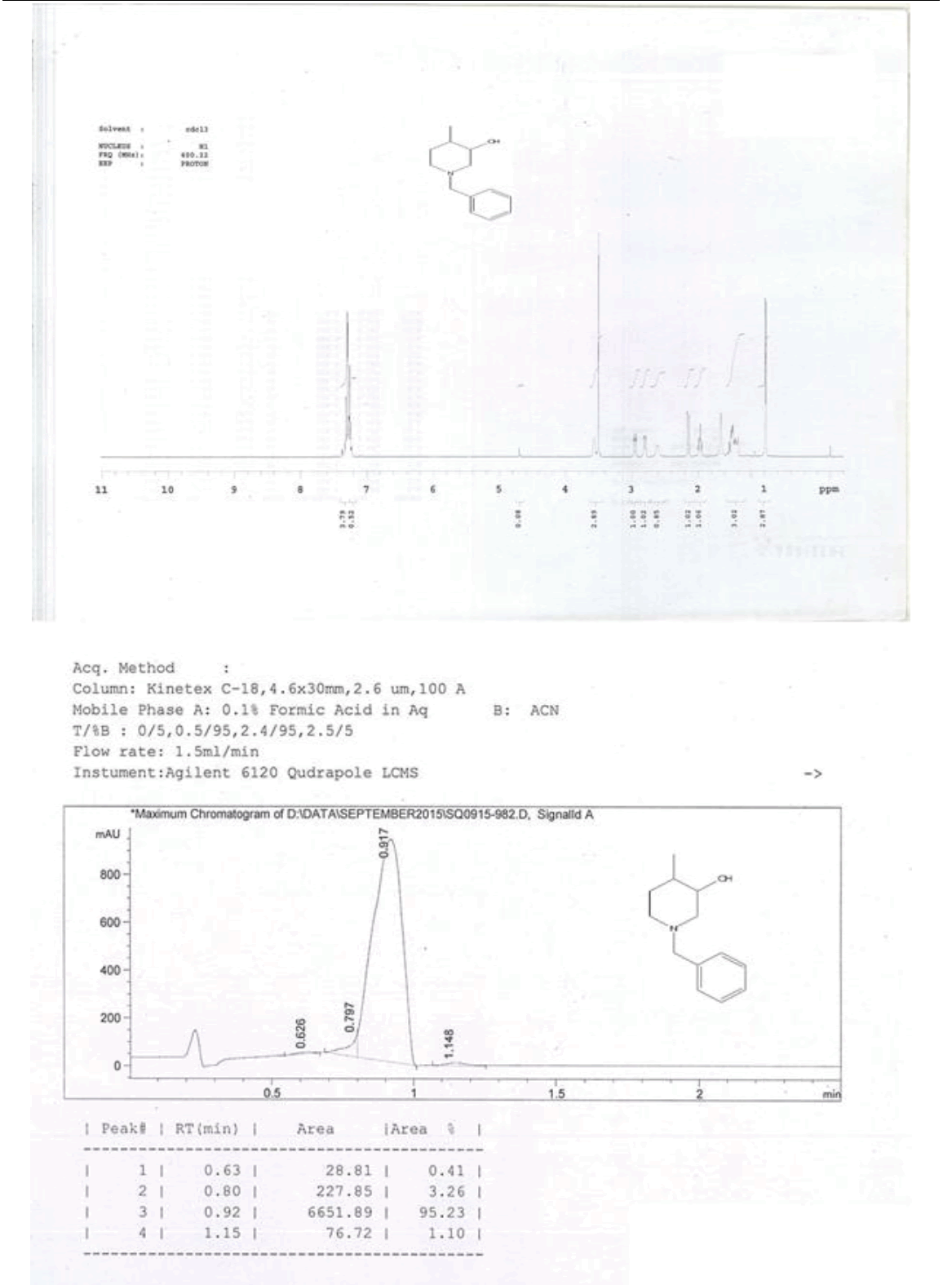
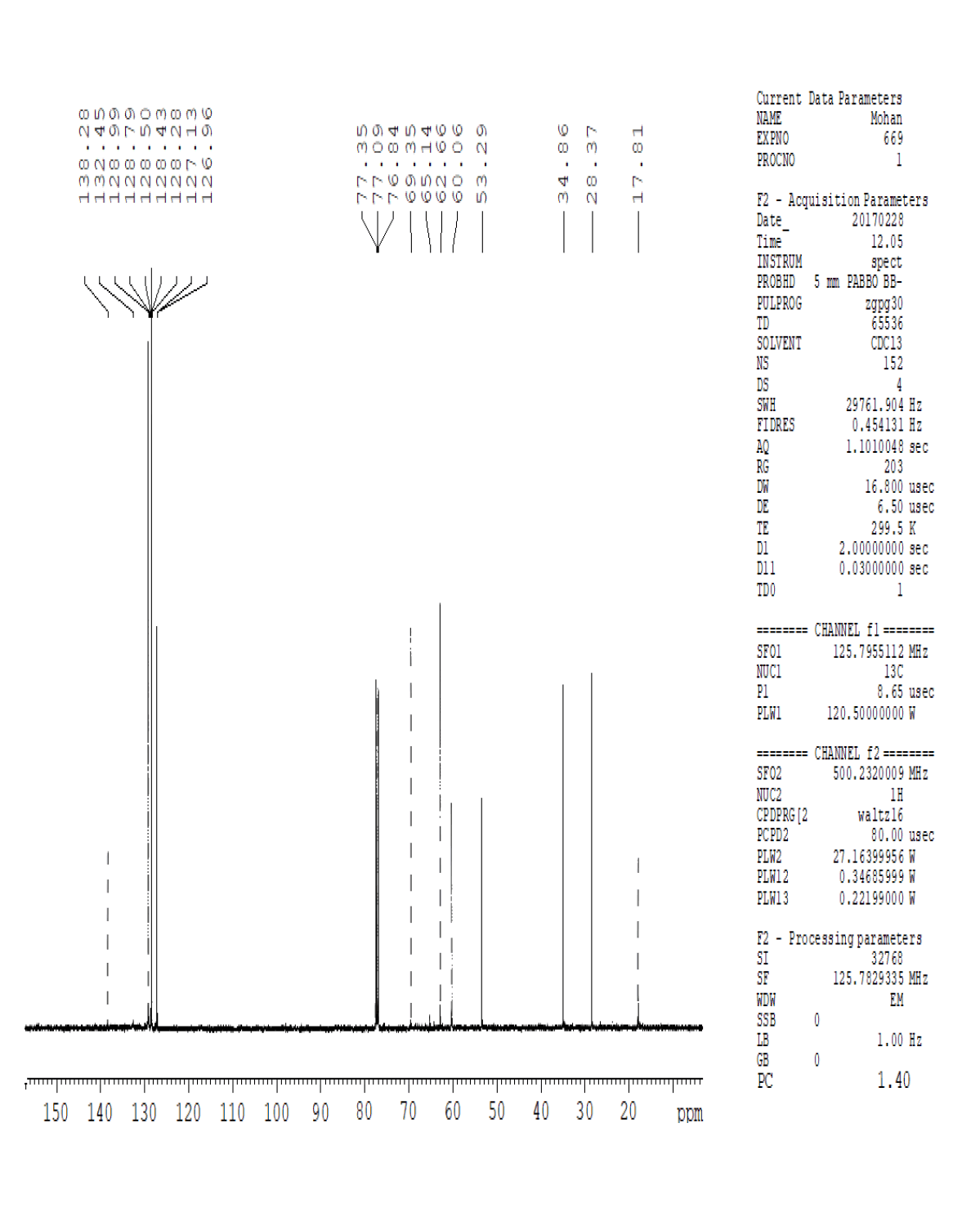
For compound 14: 1H-NMR (400 MHz, CDCl3): 7.37-7.22 (m, 5H), 3.56 (s, 1H bro), 3.50 (s, 2H), 2.96-2.81 (m, 1H), 2.72-2.62 (m, 1H), 2.60- 2.16 (m, 1H), 2.13 (m, 1H), 1.54-1.40 (m, 3H), 1.01 (s, J=6.0 Hz,3H); 13C-NMR (100 MHz, CDCl3): δ (ppm)=17.81, 28.37, 34.86, 53.29, 60.06, 60.06, 65.66, 69.35, 77.09, 127.13, 128.21, 128.90, 132.99, 138.28. LC-MS: tRet=0.92 min, M+1: 206.1.
1H, 13C-NMR and LCMS spectra
1H-NMR and LCMS of Compound 5
CMS Compound 6
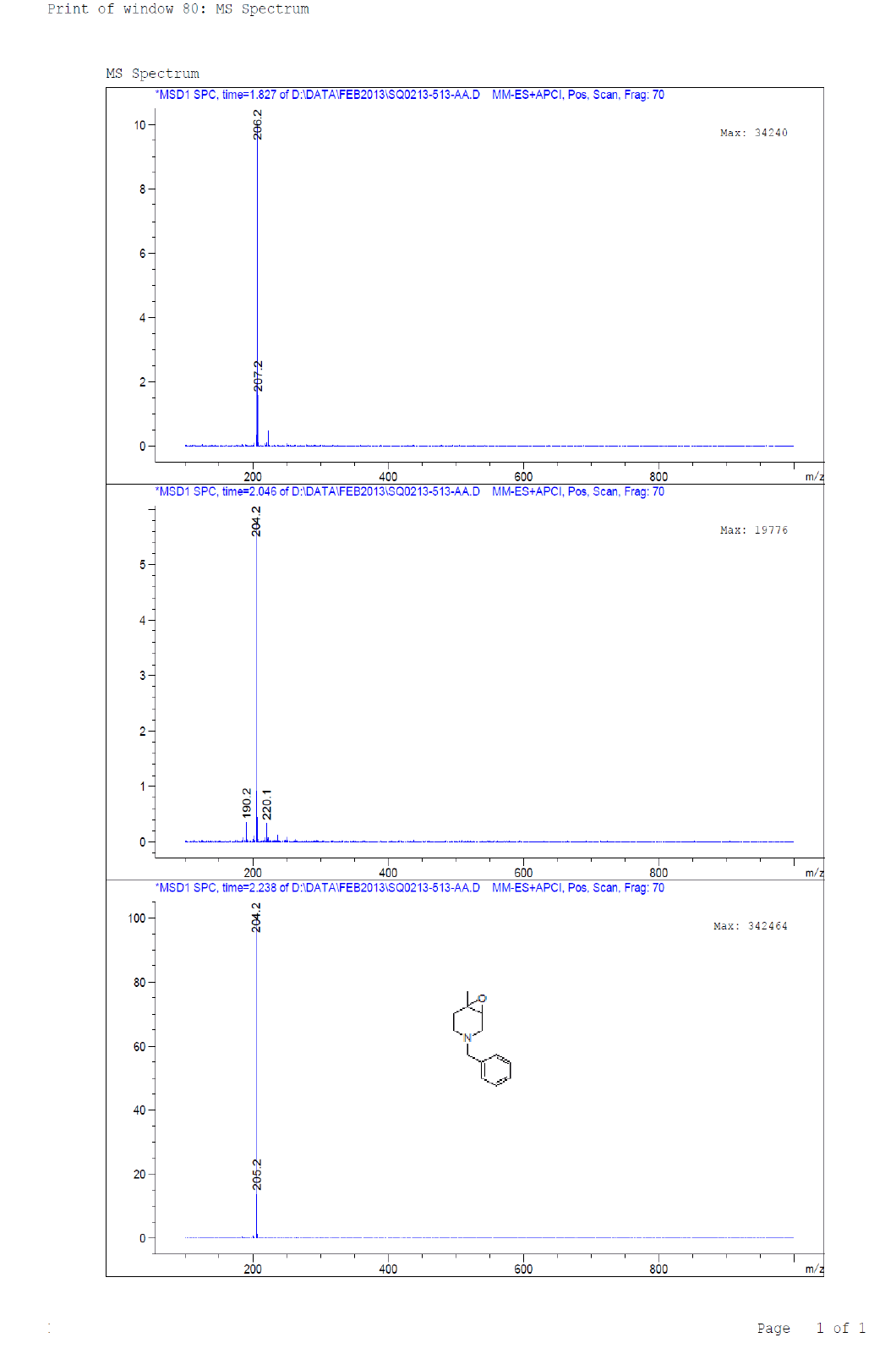
1H-NMR and LCMS of Compound 7
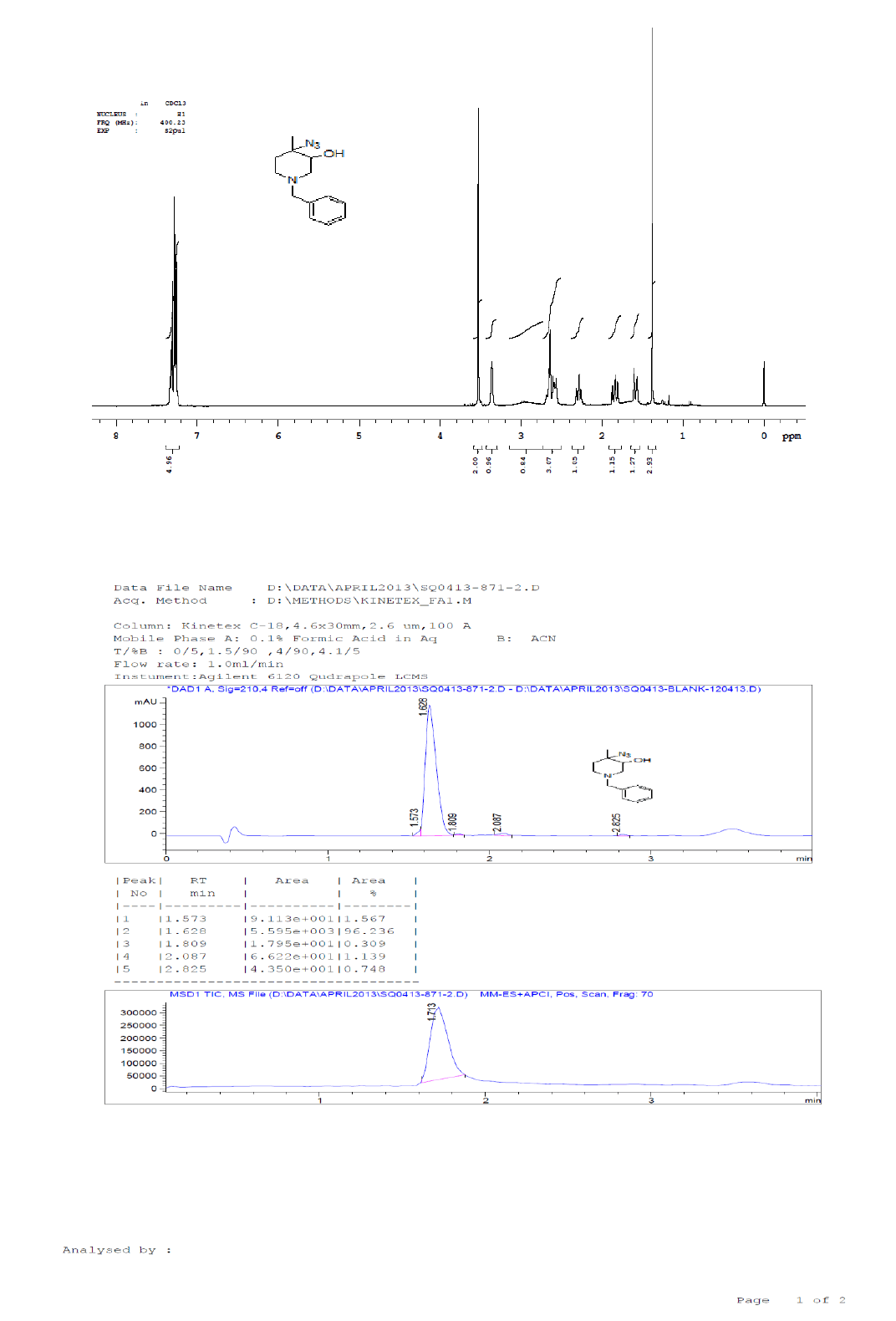
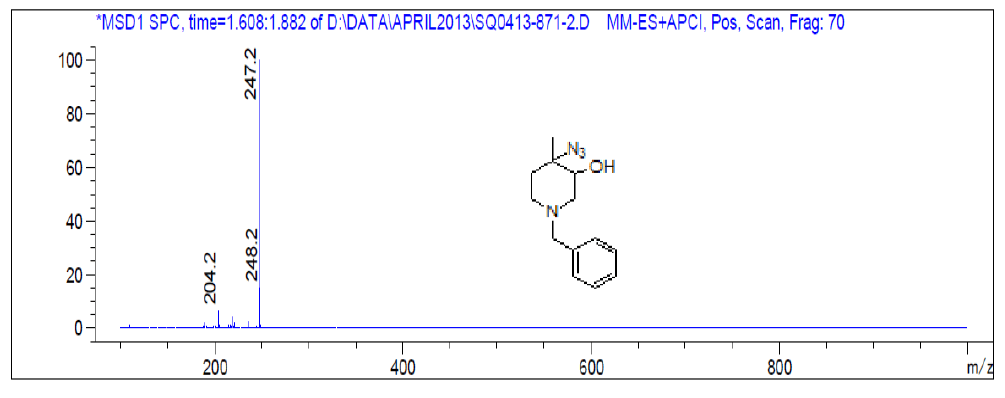
1H-NMR and LCMS of Compound 9
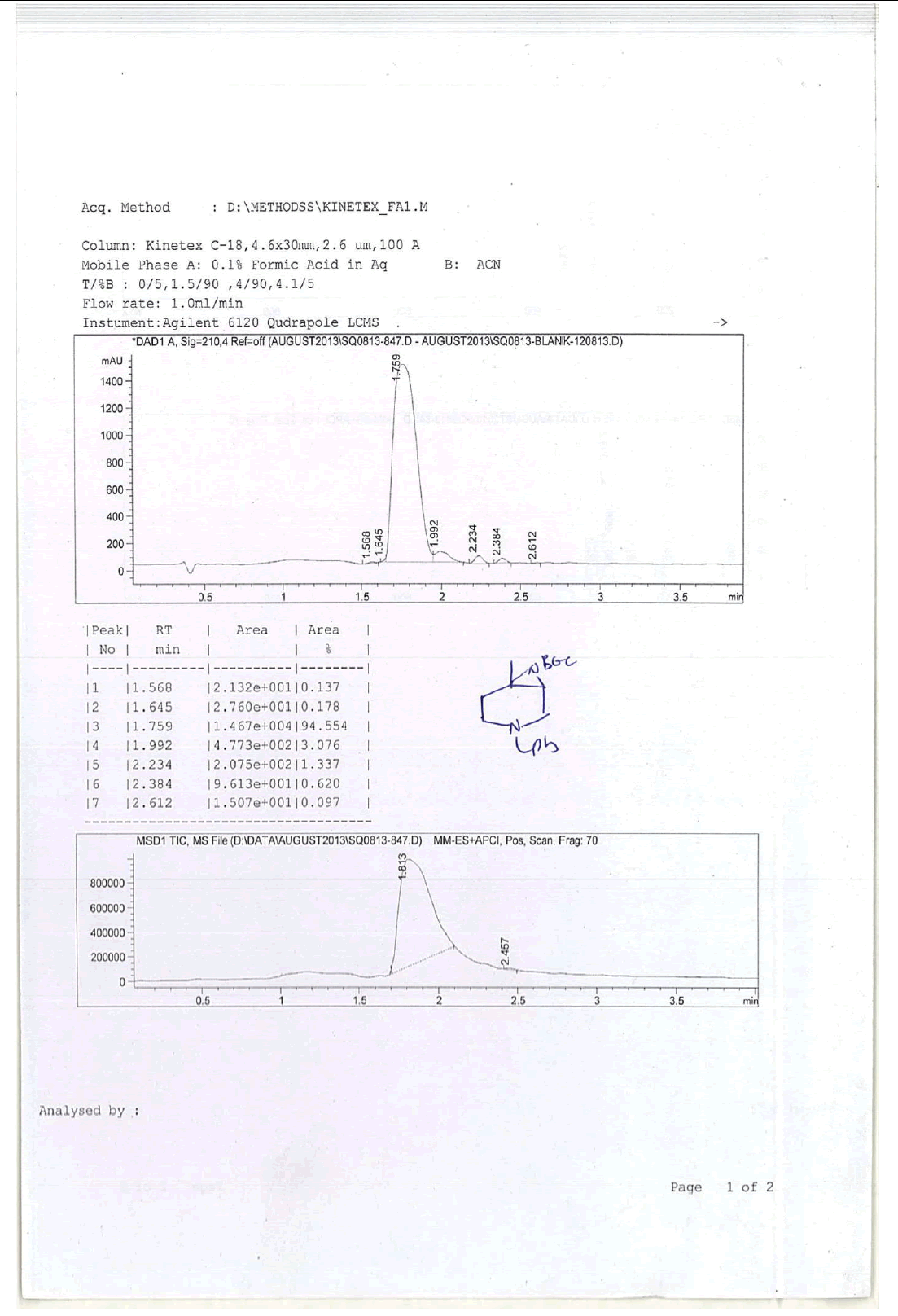
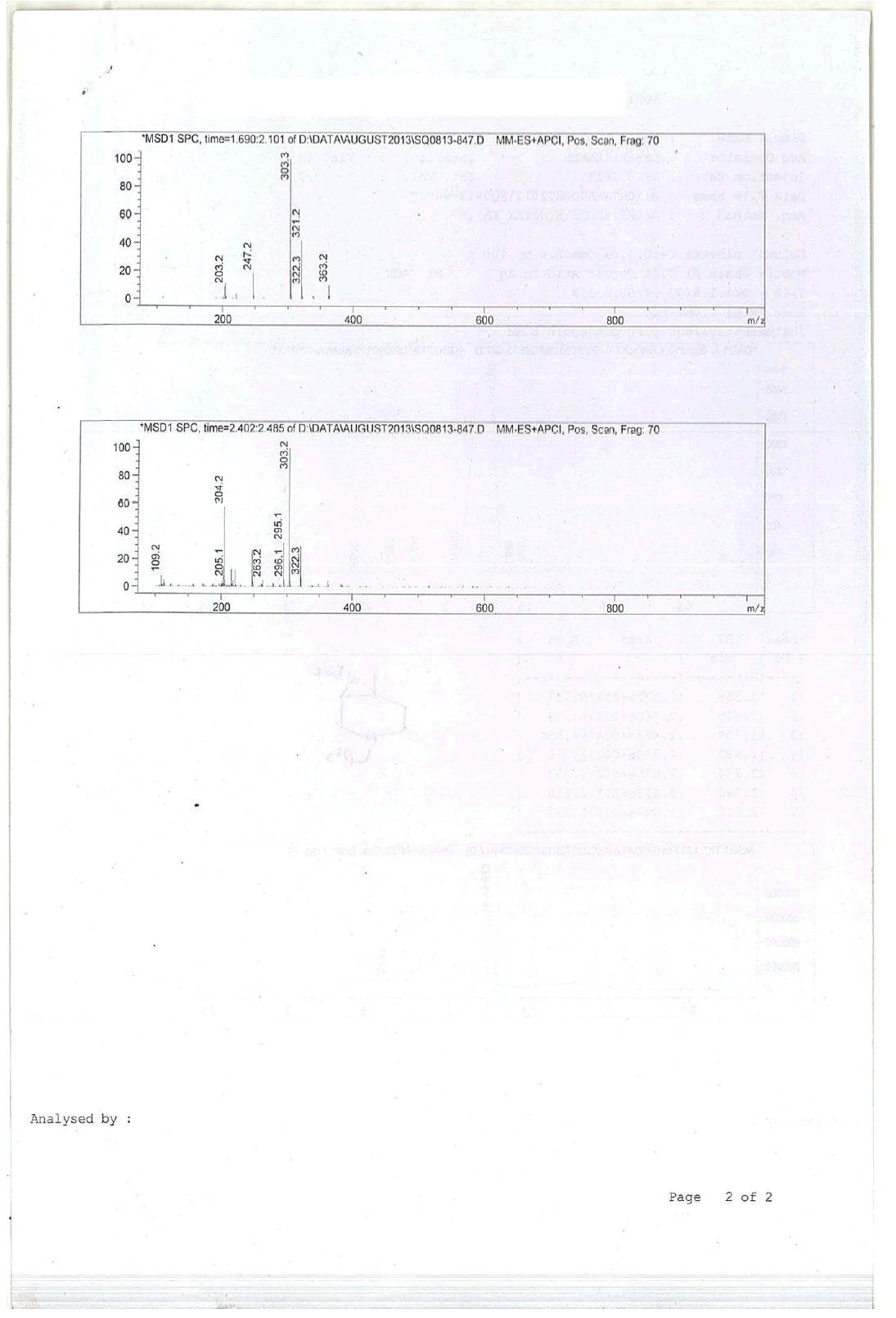
1H-NMR and HPLC of Compound 10

1H-NMR and HPLC of compound
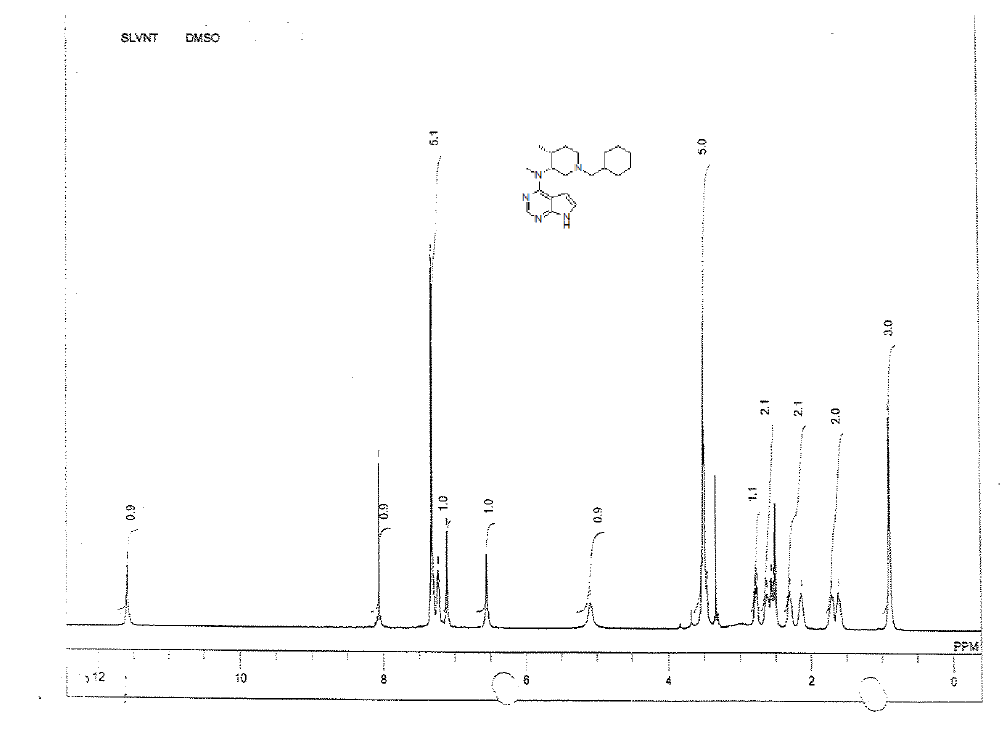
1H-NMR in DMSO, CDCl3, 13C and LCMS of compound