Research Article - Der Pharma Chemica ( 2019) Volume 11, Issue 5
Design, Synthesis and Molecular Modeling of New 1,3,5-Triazine Derivatives as Anticancer Agents
Marwa I. Serag*, Rania M. Gomaa, Mohammed A.M. Massoud and Hassan M. EisaMarwa I. Serag, Department of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt, Email: dr_marwa_1992@yahoo.com
Abstract
A new series of 1,3,5-triazine analogues were designed, synthesized and in vitro screened by the US National Cancer Institute (NCI) for their ability to inhibit 60 different human tumor cell lines. Compound 6 the most active member in this study, showing effectiveness toward numerous cell lines belonging to different tumor cell lines that reach to 60.13% inhibition in renal cancer (CAKI-1) cell line, also it has good inhibitory activity against PI3Kγ (IC50=6.90 μM) closer activity to reference wortmannin (IC50=3.19 μM). Molecular docking reveal that compound 6 occupied the same pocket of the active site of PI3Kγ and these, consistent with biological results.
Keywords
Anticancer activity, Cyanuric chloride, 1,3,5-triazine, PI3K, Molecular docking.
Introduction
Cancer or in other expression uncontrolled cellular proliferation resulted from the genetic alterations of three main types of genes; protooncogenes, tumor suppressor genes, and DNA repair genes, lies at the core of cancer as a pathological process [1]. Phosphatidylinositol 3- kinases (PI3Ks) are lipid kinases, which phosphorylate the 3-hydroxyl group of the inositol ring of phosphoinositides. As the most important phosphorylated product, phosphatidylinositol 3, 4, 5-trisphosphate (PIP3) works as a second messenger that plays leading roles in substantial cellular responses such as cell growth, survival, motility and metabolism. Alterations in the phosphatidylinositol 3-kinases (PI3K) pathway are known to play a sizable role in the buildup of cancer and provide a possible target for new therapies [2,3]. Many 1,3,5- triazines derivatives were found to be PI3K inhibitors as ZSTK474 compound I and AMG 511 compound II [4,5]. 1,3,5-triazine derivatives typify one of the most effective categories having diverse biological activity [6-11]. Altretamine III 1,3,5- triazine derivative drug that is used in treatment of ovarian cancer [12-15], also a variety of substituted 1,3,5-triazine derivatives have been synthesized and exhibited inhibitory activities against various cell lines [16,17], as compound IV (Figure 1) [18].
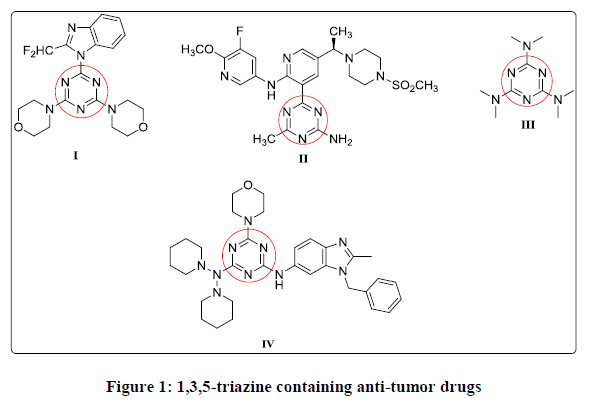
Figure 1: 1,3,5-triazine containing anti-tumor drugs
Based on the previous information, a series of new mono, di, and trisubstituted 1,3,5-triazine containing compounds were designed and synthesized. (Figure 2) 1,3,5-triazine moiety was used as a scaffold that coupled with piperidine or morpholine as heterocyclic ring or odisubstituted phenol as illustrated in Figure 2. Moreover varying the substitution pattern at 1,3,5-triazine moiety was made with either phenyl ring attached through amine linkage or ether linkage, or heterocyclic ring through amine linkage hoping to enhance the biological activity as a result of these hybridizations.
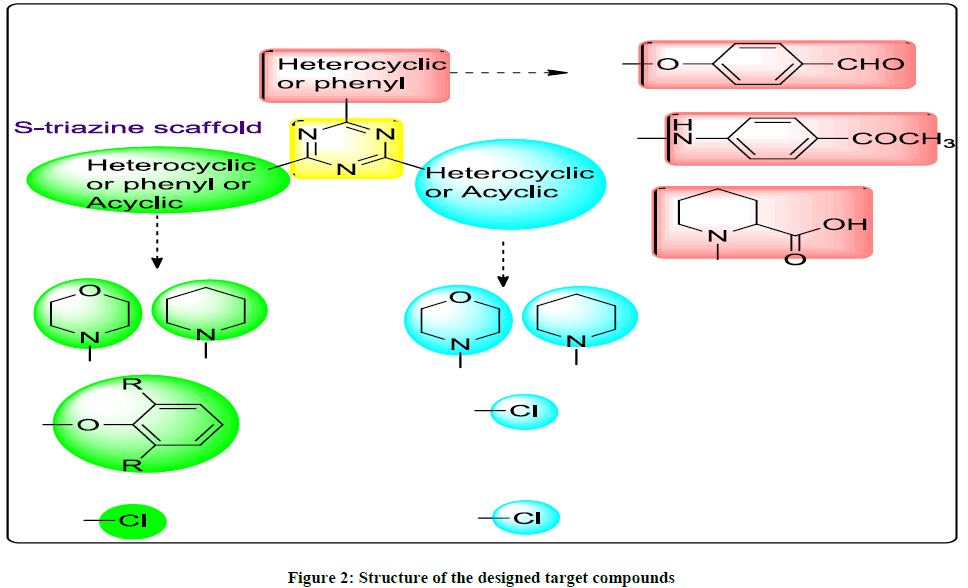
Figure 2: Structure of the designed target compounds
Materials and Methods
Chemistry
Melting points (ºC) were recorded using Fisher-John melting point apparatus and are uncorrected. Microanalyses were performed at the microanalytical unit, Cairo University. IR spectra were recorded on Mattson 5000 FT-IR spectrometer (υ in cm-1) using KBr disk. The 1H-NMR and 13C-NMR spectra were recorded on Bruker Ac 400 FT NMR spectrometer (400 MHZ), Faculty of Pharmacy, Mansoura University. The chemical shifts in ppm are expressed in δ units using tetramethylsilane (TMS) as internal standard. MS analyses were performed on JOEL JMS- 600H spectrometer in Cairo University. Reaction times were determined using TLC technique on Silica gel plates 60 F245 E. Merk, and the spots were visualized by U.V. (366 nm). Compound 1 and 4 were synthesized according to the reported method [19,20].
General procedure for preparation of compounds 2 and 3
A solution of 4-(4,6-dichloro-1,3,5-triazin-2-yloxy)benzaldehyde (1) (2.7 g, 0.01 mol) in dry DMF and amine (0.023 mol) and potassium carbonate (2.76 g, 0.02 mol) was heated at 70°C for 24 h. On completion of the reaction, the reaction mixture teeming in crumbled ice and obtained precipitate was filtered, dried and crystallized from methanol.
4-(4,6-di(piperidin-1-yl)-1,3,5-triazin-2-yloxy)benzaldehyde (2): Yield 60%; MP >300°C; IR: 1701 (C=O), 2851, 2934 (H-C=O). 1H-NMR: δ 1.45-1.46 (m, 12H, 6X (CH2- piperidine -3, -4, and -5)), 3.62-3.69 (m, 8H, 4X (CH2- piperidine -2, and -6)), 7.41 (d, J=8.5 Hz, 2H, Ar-H), 7.96 (d, J=8.5 Hz, 2H, Ar-H), 9.99 (s, 1H, CHO). MS: m/z 367 [M+], 368 [M++1). Anal. calcd for C20H25N5O2: C, 65.37; H, 6.86; N, 19.06. Found: C, 65.66; H, 6.53; N, 19.21.
4-(4,6-dimorpholino-1,3,5-triazin-2-yloxy)benzaldehyde (3): Yield 69%; MP >300°C; IR: 1711 (C=O), 2849, 2930 (H-C=O). 1H-NMR: δ 3.68-3.74 (m, 16H, 8X (CH2- morpholine H)), 7.42 (d, J=8.5 Hz, 2H, Ar-H), 7.96 (d, J=8.6 Hz, 2H, Ar-H), 9.99 (s, 1H, CHO). MS: m/z 371 [M+], 372 [M++1). Anal. calcd for C18H21N5O4: C, 58.21; H, 5.70; N, 18.86. Found: C, 58.55; H, 5.42; N, 18.77.
General procedure for preparation of compounds 5 and 6
A solution of 1-(4-(4,6-dichloro -1,3,5-triazin-2-ylamino)phenyl)ethanone (4) (2.83 g, 0.01 mol) in 1,4-dioxane, 2,6-disubstitutedphenol (0.01 mol) and potassium carbonate (1.38 g, 0.01 mol) was whiskered for 12 h at 25ºC, therewith teeming in crumbled ice. The formative precipitate filtered, washed with water and crystallized from acetone.
1-(4-(4-chloro-6-(2,6-dichlorophenoxy)-1,3,5-triazin-2-ylamino)phenyl)ethanone (5): Yield 76%; MP 232-234°C; IR: 1704 (C=O), 3500 (NH); 1H-NMR: δ 3.35 (s, 3H, CH3-CO), 7.46-7.48 (m, 3H, (dichlorophenoxy)), 7.65 (d, J=7.8 Hz, 2H, Ar-H), 7.70 (d, J=7.9 Hz, 2H, Ar-H), 10.99 (s, 1H, NH, D2O-exchangeable); 13C-NMR: 26.9, 120.1, 128.4, 128.9, 129.4, 129.7, 132.5, 142.5, 144.5, 166.3, 171.05, 171.4, 196.9. MS: m/z 409.5 [M+]. Anal. calcd for C17H11Cl3N4O2: C, 49.84; H, 2.71; N, 13.68. Found; C, 50.14; H, 2.90; N, 13.88.
1-(4-(4-chloro-6-(2,6-diisopropylphenoxy)-1,3,5-triazin-2-ylamino)phenyl)ethanone (6): Yield 70%; MP 85-87°C; IR: 1708 (C=O), 3515 (NH); 1H-NMR: δ 1.11 (d, J=6.4 Hz, 12H, 4X (CH3-diisopropyl)), 2.79-2.89 (m, 2H, 2X (CH-diisopropyl)), 3.95 (s, 3H, CH3-CO), 7.27 (s, 3H, (diisopropylphenoxy)), 7.58 (d, J=7.8 Hz, 2H, Ar-H), 7.94 (d, J=7.9 Hz, 2H, Ar-H), 10.61 (s, 1H, NH, D2O-exchangeable). MS: m/z 424.5 [M+]. Anal. calcd for C23H25ClN4O2: C, 65.01; H, 5.93; N, 13.19. Found: C, 65.31; H, 5.63; N, 13.49.
procedure for preparation of 1-(4,6-dichloro-1,3,5-triazin-2-yl)piperidine-2-carboxylic acid (7): pipecolic acid (1.29 g, 0.01 mol) in acetone was added slowly to cyanuric chloride (1.84 g, 0.01 mol) in acetone with constant stirring for 18 h in crumbled ice-cold at 0 to 5ºC. Periodically, sodium carbonate solution (0.53 g, 0.005 mol, in 10 ml water) was added dropwise to neutralized HCl evolved during the reaction, therewith the reaction mixture was teemed in crumbled ice. The formative precipitate filtered, washed with water and crystallized from tetrahydrofurane: Yield 69%, M.P >300ºC; IR cm-1: 1714 (C=O), 3424 broad (OH-C=O); 1H-NMR: (DMSO-d6), δ: 1.24-1.29 (m, 6H, 3X (CH2- piperidine -3, -4, and -5), 2.15-2.22 (m, 2H, (CH2- piperidine -6), 3.72 (t, 1H, (CH-piperidine -2), 10.68 (s, 1H, OH-C=O, D2O exchangeable). MS: m/z 277 [M+]. Anal. Calcd for C9H10Cl2N4O2: C, 39.01; H, 3.64; N, 20.22 Found: C, 39.22; H, 3.75; N, 20.13.
Biology
Cytotoxicity screening
Specifics of the procedure for the NCI 60 cell line screening are obtainable at http://dtp.nci.nih.gov/branches/btb/ivclsp.html. Substantially, for 24 h the cells were expanded in supplemented RPM1 1640 medium. The checking compound was thawed in DMSO and brood with cells at wanted concentrations. With regard to the five dose study, the compound was thawed in five concentrations with 10-fold dilutions (10-4, 10-5, 10-6, 10-7, and 10-8) M. Extension of cold trichloroacetic acid finished the assay, then the cells were fixed and soiled with sulforhodamine B. The restricted stain was thawed, and the absorbance was read by an automated plate reader. The cytostatic parameters, 50% growth inhibition (GI50), were matured from time zero, monitoring growth, and the absorbance of the five concentration levels. Inhibitory concentrations (LC50), symbolizes the average of two independent experiments. The evaluation of the compound versus the 60 humanitarian tumor cell lines with a single dose of 10 μM is done by following the same procedure as that applied for the five-dose screening. The compounds that display more than 60% of growth inhibition in at least eight tumor cell lines are only chosen for the five dose testing [21-23].
Enzyme activity inhibition assay
The kit was based on ELISA technology. Assay principle depend on that PI3 Kinase phosphorylates PI(3,4)P2 (PIP2) converting it to PI(3,4,5)P3 (PIP3). The PH domain of the protein GRP-1 set bounds to PIP3 with high affinity and specificity. This recombinant protein is included in the kit that is used as the capture protein that links to the glutathione plate and captures either the PIP3 generated as part of the kinase reaction or the biotinylated-PIP3 tracer included in the kit. Using streptavidin-HRP conjugate, the captured biotinylated-PIP3 is revealed and a colorimetric read out. The lower the signal, the higher the PI3 Kinase activity. Assay protocol state that firstly, the reaction mixture is prepared according to the manufacturer instructions with the following reagents (5X reaction buffer, PIP2 (50 μm), kinase, Wortmannin or customer compound, distilled H2O).
The PI3 Kinase reaction is setup in the Glutathione-coated strips/plate for inhibitor reaction by firstly, preincubating the kinase and inhibitor for 10 min prior to adding PIP2 substrate, then adding 5 μl/well of 5X kinase reaction buffer after that adding 5 μl/well of PIP2 substrate. Finally, adding distilled H2O to each well to make up to a final 25 μl/well. Incubate at room temperature for 1 h. 25 μl/well of Biotinylated-PIP3/EDTA working solution is added excluding the buffer control wells. 25 μl/well 1XTBS is added to the buffer control wells. 50 μl/well of GRP1 working solution is added to all wells. Incubate at chamber temperature for 1 h. The wells 4 times are rinsed with 200 μl/well 1XTBST. 50 μl/well SA-HRP working solution is added, incubate at chamber temperature for 1 h. The wells are rinsed 3 times with 200 μl of 1X TBST per well, then 2 times with 200 μl of 1X TBS per well. 100 μl of the Substrate TMB is added per well, develop in the dark for 5-20 min. Monitor the appearance of the blue color to avoid over-development. The reaction is stopped by adding 100 μl of the stop solution per well. Read at 450nm. The lower the signal, the higher the PI3 Kinase activity.
Molecular docking
Molecular modeling calculations and docking studies were carried out using molecular operating environment (MOE) software version 2014.09 (Chemical Computing Group Inc., Montreal, Quebec, Canada). The file representing the crystal structure of PI3Kγ with wortmannin was obtained from protein data bank (PDB ID: 1E7U) http://www.rscb.org. All water molecules in PDB were ignored and hydrogen atoms were added to the protein, the energy minimized using MMFF94x force field and the conformers generated were docked into the PI3Kγ receptor with MOE-DOCK using the triangle matcher placement method and the London dG scoring function. The validated docking protocol in the active site was then used to study the ligand-receptor interactions for the novel compounds to predict their binding mode and binding affinity.
Conclusion
Novel series of 1,3,5-triazine analogues were synthesized and in vitro screened by the US National Cancer Institute (NCI) for their ability to inhibit 60 different human tumor cell lines. Compound 6 displaying remarkable activity against most of the human tumor cell lines. Also, it has a PI3Kγ inhibitory activity close to that of wortmannin that match with its docking results. It is expected that the target compounds ant proliferative activity may be due to inhibition of PI3Kγ activity.
Acknowledgment
The authors are thankful to Faculty of Pharmacy, Mansoura University for funding this work and the National Cancer Institute (NCI), Bethesda, Maryland, USA, for performing the anticancer evaluation over the 60-cancer cell line panel.
References
- P. Duesberg, R. Li, Cell cycle., 2003, 2, 201-209.
- A. Akinleye, P. Avvaru, M. Furqan, J. Hematol. Oncol., 2013, 6, 88.
- L.M. Thorpe, H. Yuzugullu, J.J. Zhao, Nat. Rev. Cancer, 2015, 15, 7.
- D. Kong, T. Yamori, Curr. Med. Chem., 2009, 16, 2839-2854.
- Y. Jeong, D. Kwon, and S. Hong, Future Med. Chem, 2014, 6, 737-756.
- G. J. Kumar, H. S. Bomma, E. Srihari, med. Chem. Res., 2013, 22, 5973-5981.
- R.V. Patel, P. Kumari, D.P. Rajani, Future Med. Chem., 2012, 4, 1053-1065.
- H. Ojha, P. Gahlot, A. K. Tiwari, Chem. Biol. Drug Des., 2011, 77, 57-62.
- X. Ma, T.Y. Poon, P.T.H. Wong, Bioorg. Med. Chem. Lett., 2009, 19, 5644-5647.
- H.A. Elshemy, E.K. Abdelall, A.A. Azouz, Eur. J. Med. Chem., 2017, 127, 10-21.
- Y.Z. Xiong, F.E. Chen, J. Balzarini, Eur. J. Med. Chem., 2008, 43, 1230-1236.
- V. Talwar, V. Goel, S. Raina, Curr. Med. Res. Pract., 2016, 6, 109-112.
- M. Markman, J.A. Blessing, D. Moore, Gynecol Oncol.,1998,69, 226-229.
- M.L. Rothenberg, P. Liu, S. Wilczynski, Gynecol Oncol., 2001, 82, 317-322.
- P. Deepa, P. Kolandaivel, K. Senthilkumar, Mater. Sci. Eng. C., 2012, 32, 423-431.
- M. El-Hamamsy, N. El-Mahdy, Life Sci. J., 2014, 11, 798-805.
- A.B. Patel, K.H. Chikhalia, P. Kumari, J. Saudi Chem. Soc., 2014, 18, 646-656.
- P. Singla, V. Luxami, K. Paul, Bioorg. Med. Chem., 2015, 23, 1691-1700.
- P. Kathiriya, D. Purohit, J. Chem. Pharm. Res., 2012, 4, 383-386.
- P. Desai, K. Desai, J. Ind. Chem. Soc., 1994, 77, 155.
- P. Skehan, R. Storeng, D. Scudiero, J. Natl. Cancer Inst., 1990, 82, 1107-1112.
- M.R. Boyd, K.D. Paull, Drug Dev. Res., 1995, 34, 91-109.
- A. Monks, D. Scudiero, P. Skehan, J. Natl. Cancer Inst., 1991, 83, 757-766.
- E.H. Walker, M.E. Pacold, O. Perisic, Mol. Cell., 2000, 6, 909-919.



